Les recherches sur les collections d’échantillons biologiques humains : la place centrale des biobanques
Introduction
Les biobanques sont des établissements qui conservent et préparent des éléments biologiques humains pour les mettre à disposition des chercheurs et des industriels qui en ont besoin pour conduire leurs recherches. Les biobanques conservent également les données associées aux échantillons, comme les caractéristiques biologiques du donneur ou les données génétiques obtenues par le séquençage de l’ADN des échantillons. L’activité des biobanques est donc d’une importance capitale dans le développement des thérapies géniques et cellulaires.
Les biobanques sont soumises à un encadrement aux niveaux européen et national. Cet encadrement vise à garantir la protection des droits des donneurs ainsi que la qualité et la sécurité des éléments biologiques humains, et donc protéger la santé des receveurs, tout en favorisant la recherche et l’innovation. Au niveau de l’Union européenne, plusieurs directives et règlements, complétés par des règles de bonnes pratiques, viennent harmoniser les activités de biobanque. Néanmoins, une fragmentation des législations nationales demeure puisque certaines questions relèvent de la marge de manœuvre des États membres, tandis que d’autres questions n’ont pas été harmonisées. Nous prendrons pour illustration nationale le cas de la France.
Acteurs principaux
Les donneurs et leur famille Les donneurs doivent consentir à la collecte, au stockage, à la transformation et à la diffusion de leurs échantillons biologiques et de leurs données de santé. Ces dons sont effectués à titre gratuit : la personne ne peut pas être rémunérée mais cela n’empêche pas qu’elle soit indemnisée. Les droits des donneurs doivent être scrupuleusement respectés, notamment le devoir d’information, l’obligation d’obtenir un consentement éclairé et le droit de le retirer à tout moment, et la confidentialité des données à caractère personnel.
Les familles des donneurs peuvent être impliquées lorsque la personne n’est pas capable de consentir elle-même (par exemple en raison de son âge ou d’une maladie). Elles peuvent également être contactées si le donneur est décédé mais qu’un usage secondaire de ses échantillons est envisagé. Enfin, elles peuvent avoir un intérêt à être informées des découvertes fortuites de maladies génétiques chez les donneurs.
Les receveurs et les patients Les receveurs et les patients sont les bénéficiaires des dons ou des produits thérapeutiques issus de dons. Ils ont un intérêt direct dans la qualité et la sécurité du matériel biologique qu’ils reçoivent.
Les biobanques Les biobanques sont les établissements qui effectuent le stockage des éléments biologiques humains. Elles peuvent aussi être impliqués dans le prélèvement des échantillons, leur transformation et leur distribution. Les biobanques doivent assurer une bonne gouvernance des échantillons et des données de santé associées en assurant le respect des droits du donneur et la qualité et la sécurité des échantillons.
Les « utilisateurs » publics et privés Les chercheurs et les entreprises du secteur pharmaceutique s’appuient sur les biobanques pour se procurer les éléments biologiques humains utilisés dans le cadre de leurs recherches, y compris pour le développement de thérapies. Les acteurs formalisent le transfert de ces échantillons entre organismes par des contrats, dits Material Transfer Agreements (MTA) et Data Transfer Agreements (DTA). Ces acteurs doivent s’assurer de la traçabilité, de la qualité et de la sécurité des échantillons et des données qu’ils obtiennent afin de respecter les règles en vigueur.
Comités d’éthique Organisme indépendant habilité à se prononcer sur des aspects éthiques. Dans l’Union Européenne, l’approbation d’un comité d’éthique est requise pour démarrer un essai clinique.
Les comités d’éthique peuvent être institués par la loi ou être établis au sein des universités et des biobanques elles-mêmes. Des éléments biologiques humains ne peuvent être prélevés ou réimplanté chez un participant à une recherche que dans le cadre d’un protocole validé par un comité d’éthique. Lorsqu’un chercheur souhaite réutiliser des échantillons prélevés dans le cadre d’un projet antérieur, il peut avoir à solliciter à nouveau l’avis d’un comité d’éthique.
Autorités nationales compétentes Différentes autorités publiques (agences, ministères) sont chargées d’autoriser et de contrôler les établissements ou autres organismes, leurs activités et leurs produits.
Les procédures peuvent porter sur :
le prélèvement, le stockage, la transformation et la cession d’éléments biologiques humains,
le traitement des données,
l’utilisation de techniques de modification du génome,
l’utilisation de cellules souches etc.
Le nombre et le type de démarches à suivre varient selon la législation nationale applicable.
Certaines autorités proposent un accompagnement aux chercheurs pour faciliter leur mise en conformité avec le cadre légal. Enfin, les autorités nationales compétentes sont aussi chargées d’inspecter les établissements menant des activités relevant de leur responsabilité et de sanctionner d’éventuelles infractions.
En France, les autorités compétentes sont :
l’Agence nationale de sécurité du médicament et des produits de santé (ANSM),
l’Agence de la biomédecine (ABM),
la Commission nationale de l’informatique et des libertés (CNIL),
le Ministère de l’enseignement supérieur et de la recherche (MESR),
les Comités de protection des personnes (CPP),
les Autorités régionales de santé (ARS),
La Haute Autorité de Santé (HAS).
Définitions
Biobanques : Le terme de biobanque désigne tout établissement qui collecte, stocke et gère des échantillons biologiques ainsi que leurs données associées, afin de les céder à des tiers à des fins de recherche, médicales ou industrielles. Toutefois, il n’existe pas de définition consensuelle des biobanques tant les biobanques sont hétérogènes ; les activités inclues dans la définition peuvent donc varier. D’autres expressions sont aussi utilisées pour désigner les biobanques, comme « centre de ressources biologiques » (CRB), « collection d’échantillons biologiques humains » ou « biothèque ».
Définition en droit international et européen En droit international, la Déclaration de Taipei définit une biobanque comme « une collection de matériel biologique avec des données s’y rapportant ». Deux éléments importent donc:
La conservation d’échantillons biologiques (cellules, tissus, sang).
La conservation des données qui y sont associées, notamment les données identifiantes ou génétiques.
Au niveau européen, l’OCDE définit les centres de ressources biologiques : « Biological resource centres are an essential part of the infrastructure underpinning life sciences and biotechnology. They consist of service providers and repositories of the living cells, genomes of organism, and information relating to heredity and the functions of biological systems. BRCs contain collections of culturable organisms (e.g. micro-organisms, plant, animal and human cells), replicable parts of these (e.g. genomes, plasmids, viruses, cDNAs), viable but not yet culturable organisms, cells and tissues, as well as databases containing molecular, physiological and structural information relevant to these collections and related bioinformatics. » (OECD, Biological Resource Centres: Underpinning the Future of Life Sciences and Biotechnologyy, 2001, p.7).
En droit de l’Union européenne, le terme de « biobanque » n’est pas explicitement défini. Pourtant, plusieurs législations peuvent s’appliquer.
D’une part, il s’agit des législations applicables à la conservation d’échantillons biologiques humains destinés à des applications humaines. Elles couvent donc les utilisations médicales de ces échantillons mais aussi leurs utilisations en recherche clinique.
La Directive 2004/23/CE du 31 mars 2004 relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains (Directive T&C) prévoit des règles pour les « établissement de cellules/tissus » qui conservent et stockent des cellules et tissues d’origine humaine destinés à des applications humaines.
À noter : cette directive va être remplacée par le « Règlement SoHO » à partir du 7 août 2027.
La Directive 2002/98/CE du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins prévoit des règles pour les « établissement de transfusion sanguine » qui conservent et stockent du sang et des composants sanguins destinés à la transfusion.
À noter : cette directive va être remplacée par le « Règlement SoHO » à partir du 7 août 2027.
Le Règlement (UE) 2024/1938 du 13 juin 2024 concernant les normes de qualité et de sécurité des substances d’origine humaine destinées à une application humaine (Règlement SoHO) prévoit des règles pour les « entités SoHO », et plus précisément ici les « établissements SoHO » qui stockent des échantillons d’origine humaine dits ‘substances d’origine humaine’.
Une « entité SoHO » exerce une ou plusieurs activités liées aux SoHO ayant une incidence directe sur la qualité, la sécurité ou l’efficacité de ces substances, comme l’enregistrement des donneurs, le prélèvement de l’échantillon, sa transformation, les procédures de contrôle de qualité, de stockage, de libération, de distribution, d’importation/exportation, ainsi que l’application humaine etc. (article 3. 33 Règlement SoHO).
Un « établissement SoHO » exerce une ou plusieurs activités de transformation, de stockage, de libération, ou d’importation/exportation de substances d’origine humaine (article 3. 35 Règlement SoHO).
Dans ce contexte l’établissement principale est défini :
« "établissement principal"
a) en ce qui concerne un responsable du traitement établi dans plusieurs États membres, le lieu de son administration centrale dans l'Union, à moins que les décisions quant aux finalités et aux moyens du traitement de données à caractère personnel soient prises dans un autre établissement du responsable du traitement dans l'Union et que ce dernier établissement a le pouvoir de faire appliquer ces décisions, auquel cas l'établissement ayant pris de telles décisions est considéré comme l'établissement principal;
b) en ce qui concerne un sous-traitant établi dans plusieurs États membres, le lieu de son administration centrale dans l'Union ou, si ce sous-traitant ne dispose pas d'une administration centrale dans l'Union, l'établissement du sous-traitant dans l'Union où se déroule l'essentiel des activités de traitement effectuées dans le cadre des activités d'un établissement du sous-traitant, dans la mesure où le sous-traitant est soumis à des obligations spécifiques en vertu du présent règlement; » (Article 4.16 du Règlement (UE) 2016/679).
Définitions en droit français La définition des biobanques varie selon le pays considéré. En France, le Code de la Santé publique (CSP) ne définit par le terme de « biobanque ». Il encadre plutôt les collections d’échantillons biologiques humains, définies comme :
« la réunion, à des fins scientifiques, de prélèvements biologiques effectués sur un groupe de personnes identifiées et sélectionnées en fonction des caractéristiques cliniques ou biologiques d'un ou plusieurs membres du groupe, ainsi que des dérivés de ces prélèvements » (Article L1243-3 CSP).
Le droit français distingue ensuite deux possibilités pour les organismes qui conservent et préparent ces collections d’échantillons :
Les organismes qui conservent et préparent des éléments biologiques pour les besoins de leurs propres programmes de recherche(Article L1243-3 CSP).
Les organismes qui conservent et préparent des éléments biologiques en vue de leur cession(Article L1243-4 CSP). C’est cette seconde qualification qui s’applique généralement aux activités des biobanques.
Définition d’un groupe d’experts auprès de la Commission européenne (en anglais) « Biobanks typically:
(a) collect and store biological materials that are annotated not only with medical, but often also epidemiological data (e.g. environmental exposures, lifestyle/occupational information);
(b) are not static “projects”, since biological materials and data are usually collected on a continuous or long-term basis;
(c) are associated with current (defined) and/or future (not yet specified) research projects at the time of biospecimen collection;
(d) apply coding or anonymisation to assure donor privacy but have, under specific conditions, provisions that participants remain re-identifiable in order to provide clinically relevant information back to the donor; and
(e) include established governance structures (e.g. ethics review committees) and procedures (e.g. consent) that serve to protect donors’ rights and stakeholder interests » (Direction Générale de la Recherche et de innovation, « Biobanks for Europe – A challenge for governance », Commission Européenne, Office des publications de l’Union européenne, 2012, p. 13).
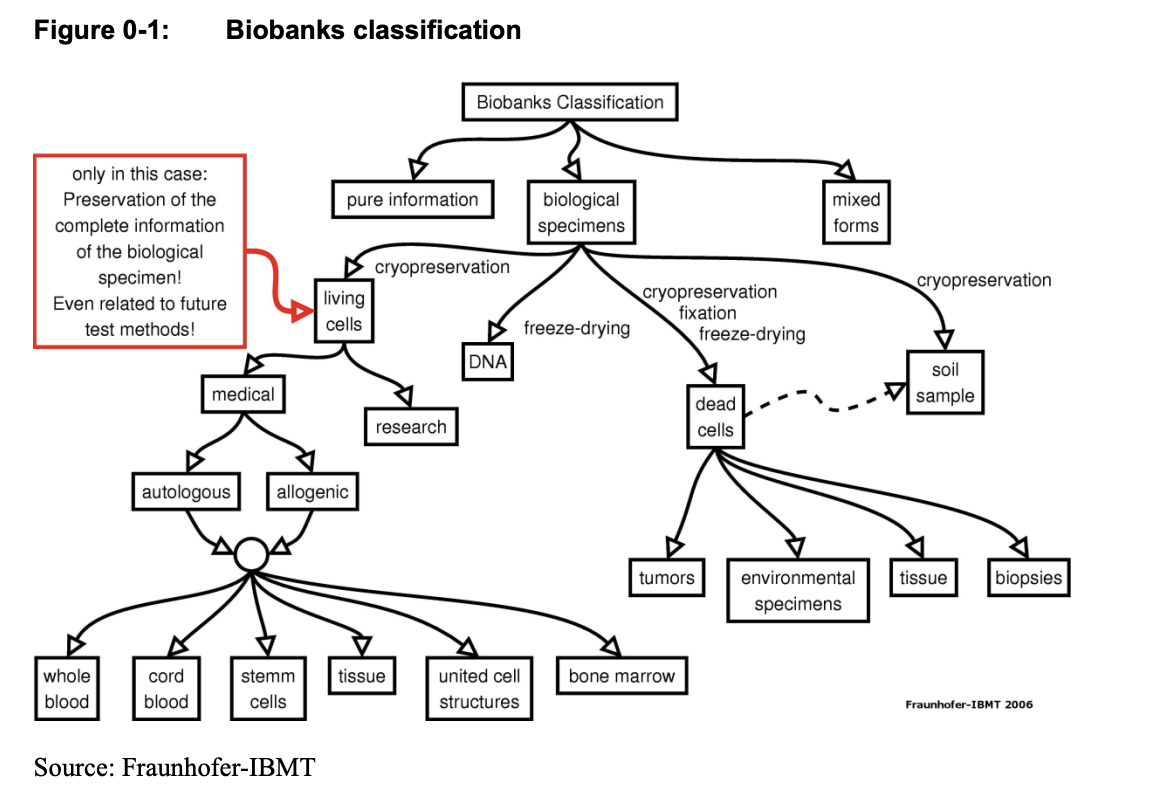
Typologie des biobanques Il existe plusieurs types de collections d’échantillons en fonction des activités qu’elles alimentent :
Les Banques de Recherche (Research Cell Bank ou RCB en anglais) sont constituées uniquement à des fins de recherche et de développement. Elles ne se conforment pas nécessairement aux normes de Bonnes Pratiques de Fabrication (BPF).
Les Banques de Cellules Maîtresses(BMC) (Master Cell Banks ou MCB en anglais) sont conformes aux normes BPF, elles assurent l’identité et la pureté des lignées cellulaires en réalisant différents tests de contrôle.
Les Banques de Cellules de Travail (BCT) (Working Cell Banks ou WCB en anglais) sont issues des BMC, elles permettent un approvisionnement régulier de cellules pour la recherche ou l’industrie.
Les Banques de Fin de Production (End-of Production Cell Bank ou EoPCB en anglas) sont constituées à partir des BCT à la fin de cycles de production. Elles permettent des contrôles de qualité en vérifiant l’absence de contaminants ou de variation dans la lignée cellulaire.
Enfin, il existe plusieurs typologies des biobanques. On en mentionnera deux ci-dessous.
Le rapport de 2012 du groupe d’experts auprès de la Commission européenne classe les biobanques en fonction de leur forme (design) et de leur objectif scientifique :
Population-based biobanks
Disease-oriented biobanks
Case-control biobanks
Tissue banks
Biobanking within the context of clinical trials
Other specific biobanking formats: Guthrie cards, cord blood, stem cells
Un rapport de 2011 auprès du Joint Research Center de la Commission européenne, classe les biobanques selon le type de matériel qu'elles conservent (ADN, tissus, cellules vivantes, données associées ou toute combinaison de ces éléments) et leur finalité (usage thérapeutique, recherche, usage clinique, etc.).
Au-delà des défis pratiques et organisationnels liés à leur fonctionnement, les biobanques sont confrontées à des enjeux d’ordre légal, éthique, économique et social, qui ont une influence directe sur le développement et l’acceptabilité de leurs opérations.
Enjeux juridiques : protection des droits du donneur dans un cadre réglementaire fragmenté
Les biobanques doivent assurer le respect des droits du donneur. Pour ce faire, les collectes d’échantillons biologiques doivent respecter les réglementations en vigueur dans chaque pays. Cela implique notamment :
Une information claire et préalable du donneur quant aux usages prévus de ses échantillons,
Un consentement éclairé,
La possibilité de retirer son consentement, d’obtenir la destruction des données, voire des échantillons détenus par la biobanque.
Un autre défi réside dans l’absence d’harmonisation de certaines questions au niveau européen. La grande variabilité des législations nationales complexifie les collaborations internationales. Des divergences concernent :
l’étendue de l’information à transmettre au donneur,
les modalités de recueil du consentement et de son éventuel retrait,
la possibilité d’usages secondaires des échantillons,
la traçabilité des échantillons,
leur éventuelle appropriation.
Dans ce contexte de fragmentation réglementaire, le travail d’harmonisation se réalise principalement grâce aux normes techniques.
Enjeux éthiques : concilier les différents intérêts collectifs et individuels
L’enjeu d’un point de vue éthique est de parvenir à concilier les différents intérêts individuels et collectifs en présence.
Le donneur a droit au respect de sa vie privée et de son autonomie. Une anonymisation totale des échantillons assure certes le respect de la vie privée du donneur, mais cela lui empêchera aussi de connaître les usages secondaires faits de ses échantillons. Cela le prive également de retours d’informations concernant d’éventuelles pathologies qui seraient détectées en utilisant ses échantillons. Il peut donc être important de maintenir un lien entre le donneur et ses échantillons.
Les patients doivent pouvoir bénéficier des thérapies innovantes dont ils ont besoin tout en bénéficiant des garanties nécessaires pour la protection de leur santé.
Les scientifiques jouissent de la liberté de recherche. Accéder aux ressources biologiques leur permet d’accroitre les connaissances humaines et d’améliorer la santé humaine. Toutefois, des limites éthiques peuvent être opposées à certains usages d’éléments biologiques humains. En outre, ces nouvelles connaissances génèrent beaucoup d’espoirs et de promesses, ce qui nécessite de communiquer de manière responsable sur les résultats de recherche.
La société dans son ensemble peut accorder une importance à la protection du corps humain, empêchant sa marchandisation ou des atteintes à la dignité humaine, tout en souhaitant le développement et l’accès aux innovations thérapeutiques.
Si ces différents intérêts ne sont pas nécessairement contradictoires, leur conciliation reste un exercice délicat. Il est alors nécessaire de combiner les discussions éthiques et le droit pour organiser les pratiques de biobanques pour qu’elles soient acceptées par les citoyens.
Enjeux économiques : assurer la viabilité des opérations
L’autonomie financière des biobanques est un enjeu crucial, en particulier lorsque celles-ci sont publiques. Les biobanques doivent amortir leurs investissements et assurer le financement durable de leurs activités. Ces impératifs doivent être conciliés avec les principes applicables aux éléments biologiques humains dans le pays considéré.
En France, par exemple, le principe de non-patrimonialité du corps humain, de ses éléments et produits interdit de les faire figurer dans le patrimoine de la personne source au titre d’un droit évaluable en argent. De même, le droit de l’Union européenne prévoit le principe des dons volontaires et non rémunérés des cellules et tissus, et plus largement des substances d’origine humaine, même si l’indemnisation des donneurs peut être autorisée par les Etats, sous réserve de garantir la neutralité financière du don (l’absence de gains ou de pertes financières résultant du don). Toutefois, les biobanques peuvent évaluer, et éventuellement facturer, le coût de leur travail sur ou pour ces échantillons, notamment s’agissant de leur conservation. Ceci soulève donc d’importantes questions concernant la tarification des activités des biobanques.
Enjeux sociétaux : gouvernance responsable et confiance du public
Les biobanques sont cruciales pour la recherche biomédicale et le développement de thérapies innovantes. Les échantillons biologiques ont en effet une réelle valeur sociale en ce que leurs utilisations participent à l’amélioration des connaissances scientifiques et la protection de la santé publique. Toutefois, ces recherches reposent sur l’adhésion et la confiance des donneurs, sans lesquels elles ne pourraient être conduites. Dans un modèle fondé sur le don gratuit, l’engagement des donneurs repose sur l’altruisme et la solidarité. Il est donc primordial que les donneurs puissent faire confiance aux opérateurs de biobanques à qui ils confient leurs échantillons. Cela se concrétise par une gestion rigoureuse et transparente des échantillons. Par exemple, des procédures peuvent être mises en place pour permettre aux donneurs de suivre les usages faits de leurs échantillons.
Opportunités et incitations
Les biobanques bénéficient de plusieurs soutiens financiers et réglementaires aux niveaux européen et nationaux.
Au niveau de l’Union européenne
L’Union européenne finance de nombreux projets liés aux biobanques dans le cadre du programme Horizon Europe. L’un d’entre eux, la BioMolecular Resources Research Infrastructure - European Research Infrastructure Consortium (BBMRI-ERIC) œuvre pour la création d’une infrastructure de recherche paneuropéenne concernant les biobanques et les ressources biomoléculaires. De nombreux outils et informations sont disponibles sur son site internet, y compris des formations et des opportunités de partenariats.
Au niveau de la France
Différents dispositifs financent les activités de recherche biomédicale. Le Ministère de l’enseignement supérieur et de la recherche pilote plusieurs programmes tels que le crédit d'impôt recherche (CIR), la stratégie nationale des infrastructures de recherche, les concours d'innovation (i-Lab, i-PhD, i-Nov), et le dispositif France 2030. Dans le cadre de France 2030, plusieurs projets spécifiquement liés aux biobanques sont financés :
France Biobank Network(FrBioNet) a pour objectif de valoriser les collections biologiques françaises et structurer le pilotage des biobanques.
Biobanque Cohortes Françaises(BioCF) vise à créer une ressource nationale centralisée pour la collecte, le stockage et l'exploitation d'échantillons biologiques issus de cohortes généralistes en population française.
Au niveau de la politique sectorielle, une initiative de structuration des opérateurs de biobanques a été formalisée dans le contrat du comité stratégique de la filière Industrie et Technologies de Santé (CSF-ITS). En outre, il existe un Groupement d’intérêt scientifique Infrastructure en Biologie Santé et Agronomie (GIS IBiSA) qui labelise et soutient les structures, accompagne les démarches qualité en aidant à la certification ISO et développe des réseaux de compétences. Le Réseau national des CRB de centres hospitalo-universitairesaideà la structuration nationale de ces CRB grâce notamment à des activités de promotion des activités de CRB, de standardisation et d’harmonisation des pratiques, et de fourniture d’expertise auprès des tutelles. Enfin, d’autres initiatives visent des besoins plus spécifiques comme le réseau national de production et de biobanques d’organoïdes (Ribbon), qui a pour objectif de faciliter la mise en place de collaborations entre plateformes de production, biobanques et chercheurs à l’aide d’un annuaire des modèles organoïdes.
Interactions avec les régulateurs
Au niveau de l’Union européenne
Il n’existe pas de possibilités d’interaction spécifique dédiée aux activités de biobanques avec les institutions ou agences européennes. C’est BBMRI-ERIC qui est l’infrastructure de référence au niveau européen. Ses différents services peuvent être contactés via leur page de contact.
Au niveau de la France
Plusieurs mécanismes permettent d’interagir avec les instances réglementaires :
Le guichet de l’Agence Innovation en Santé (AIS) permet de présenter des projets innovants et d’être redirigé vers les dispositifs d’accompagnement et de financement adaptés.
Le guichet Innovation et Orientation (GIO) de l’ANSM contribue au développement de produits de santé innovants, comme de nouveaux médicaments ou des dispositifs médicaux. Le GIO peut répondre à des questions réglementaires, aider à qualifier la recherche et le lieu de recherche etc.
En plus de ces dispositifs, il est possible de prendre contact avec différentes autorités nationales compétentes :
Les étapes pratiques qui doivent être entreprises pour mettre en conformité les activités d’une biobanque dépendront du pays considéré. Généralement :
Lorsque les prélèvements d’échantillons se font dans le cadre d’une recherche sur la personne humaine, l’ approbation d’un comité d’éthique est requise.
Le traitement des données de santé doit faire l’objet d’une déclaration ou d’une demande d’autorisation auprès de l’autorité nationale de protection des données. Des méthodologies de référence peuvent être mises à disposition par l’autorité nationale compétente.
L’établissement doit généralement être autorisé par une autorité nationale compétente, qui pourra aussi procéder à des inspections.
Certains pays disposent d’une législation spécifique aux biobanques qui unifie les procédures relatives aux échantillons et celles relatives aux données. Des autorités avec des compétences complémentaires peuvent intervenir, par exemple en fonction du type d’échantillons concerné ou de l’usage qui en est fait.
Les étapes pratiques en France
On peut distinguer les procédures relatives aux échantillons en eux-mêmes de la procédure relative à la gestion des données qui y sont associées (bien que ces deux procédures puissent être conjointes). Des procédures supplémentaires s’appliquent pour certains types d’échantillons présentant une sensibilité éthique particulière, notamment ceux qui sont modifiés génétiquement.
Obtention des échantillons :
Échantillons prélevés pour la première fois : cela concerne les échantillons que l’on souhaite obtenir dans le cadre d’une recherche impliquant la personne humaine (RIPH). L’article L1121-1 du CSP prévoit trois catégories de RIPH selon le niveau de risque pour les personnes se prêtant à la recherche : les recherches interventionnelles qui comportent une intervention sur la personne non justifiée par sa prise en charge habituelle (RIPH 1), les recherches interventionnelles à risques et contraintes minimes (RIPH 2) et les recherches non interventionnelles (RIPH 3). Cette procédure est généralement effectuée par les chercheurs ou les plateformes de production plutôt que les biobanques lorsqu’il s’agit d’entités distinctes.
Chaque projet RIPH doit être enregistré avant la transmission des documents aux différentes autorités compétentes. Les essais cliniques de médicaments couverts par le Règlement UE 2014/536 du 16 avril 2014 relatif aux essais cliniques de médicaments à usage humain devront posséder un numéro EudraCT. Les RIPH 1 devront obtenir un numéro IDRCB auprès de l’ANSM.
Une RIPH doit obtenir un avis favorable d’un comité de protection des personnes (CPP) (Article L1123-14 CSP). Le CPP examine notamment le respect des droits des personnes, les procédures d’information et de recueil du consentement éclairé, la nécessité et la proportionnalité des activités prévues au regard des finalités de la recherche. L’attribution de la demande à un CPP se fait au hasard (Article L1123-14 CSP). Le CPP se prononce dans un délai de 45 jours (Article R1123-23 CSP), son silence valant refus. La procédure se fait sur le site CNRIPH. À noter : s’il est envisagé de conserver les échantillons après la fin de la recherche, les donneurs doivent en être informés lors de la procédure d’information pour qu’ils puissent y consentir. À défaut, les promoteurs devront repasser par un CPP à la fin de la recherche pour valider ce nouvel usage.
L’Agence nationale de sécurité du médicament et des produits de santé (ANSM) sera impliquée pour toute procédure de RIPH. Pour les RIPH 1, l’autorisation de l’ANSM est requise (Article L1123-12 CSP). Si la recherche a lieu dans un établissement de santé, l’Agence régionale de santé doit aussi autoriser le lieu (Article L1121-13 CSP). Pour les RIPH 2 et 3, le promoteur doit transmettre à l'ANSM l'avis final rendu par le CPP ainsi que le résumé de la recherche sur le site démarches-simplifiées (Article L1121-4 CSP).
Échantillons déjà prélevés : cela concerne les échantillons déjà obtenus dans une RIPH antérieure ou lors d’une activité de soin. Il peut s’agir de mises en commun des résidus de chirurgies à des fins de recherches ou d’usages secondaires d’échantillons initialement prélevés dans le cadre d’une RIPH. Cette procédure est donc particulièrement destinée aux activités de stockage réalisées par une biobanque.
Le Ministère de l’enseignement supérieur et de la recherche (MESR) reçoit les déclarations des établissements qui souhaitent conserver et préparer le matériel biologique humain pour leurs propres programmes de recherche (Articles L1243-3 et R1243-49 à R1243-56 CSP). En revanche, lorsqu’un établissement a pour objectif de céder ce matériel à des tierces personnes à des fins scientifiques, une autorisation est requise (Articles L1243-4 et R1243-57 à R1243-66 CSP). En outre, lorsque l’organisme est un établissement de santé, la déclaration ou la demande d’autorisation sont également adressées à l’Agence régionale de santé. Pour les déclarations, le silence de l’administration après deux mois vaut accord. Pour les demandes d’autorisations, la procédure d’instruction de la demande dure trois mois et le silence de l’administration vaut rejet. Les autorisations sont valables pour cinq ans et peuvent être renouvelées avec présentation d’un rapport d’activité (Article R1243-63 CSP). Les procédures se font via le portail CODECOH. Les biobanques sont donc soumises à une autorisation du MESR lorsqu’elles veulent réaliser des mises à dispositions à des utilisateurs extérieurs, mais peuvent opérer avec une déclaration au MESR lorsqu’elles ne font que conserver et céder du matériel biologique aux équipes de leur organisme.
Le MESR autorise également les activités d’importation et d’exportation des éléments biologiques humains à des fins scientifiques (Article L1211-1 CSP). Le MESR vérifie alors la gratuité du don, le consentement éclairé et le respect des règles d’étiquetage et de d’emballage. Cette autorisation est délivrée sous trois mois.
Si une collection provient d’une activité de soins ou d’une RIPH sans consentement initial pour une conservation prolongé, un CPP doit intervenir pour la requalifier en collection de recherche. La procédure est le même que pour une RIPH et se réalise sur le site CNRIPH.
Données de santé associées aux échantillons : la Commission Nationale Informatique et Libertés (CNIL) met à disposition des méthodologies de référence qui permettent aux prometteurs de faire un engagement de conformité. Pour les RIPH, les MR-001 ou MR-003 sont applicables, tandis que les recherches hors RIPH devront recourir au MR-004. Pour les biobanques, c’est le référentiel « entrepôt de données de santé » qui sera applicable. Si une activité n’entre pas dans une des méthodologies de référence (Article 66(III), Loi Informatique et Libertés) mais qu’elle a une finalité d'intérêt public de recherche, d'étude ou d'évaluation dans le domaine de la santé (Article 54 Loi Informatique et Libertés), une demande d’autorisation doit être adressée à la CNIL. Les déclarations de conformités se font sur le portail dédié de la CNIL et les demandes d’autorisations se font sur le site démarches-simplifiée.
Des procédures additionnelles sont applicables dans les cas suivants :
Échantillons éthiquement sensibles : l’utilisation de certains éléments biologiques humains requièrent une déclaration ou une demande d’autorisation à l’Agence de la biomédecine (ABM).
Les recherches nécessitant le prélèvement d’organes, de tissus ou de cellules sur une personne décédée, ou le prélèvement de tissus ou de cellules embryonnaires ou fœtaux à l’issue d’une interruption de grossesse, doivent être déclarées à l’ABM (Articles L1232-3, L1241-5 et L1241-6 CSP). L’absence de réponse sous deux mois vaut accord. Plus d’informations sur cette procédure sur le site de l’ABM ici.
Les établissements souhaitant conserver des embryons à des fins de recherche doivent demander une autorisation à l’ABM. Les établissements autorisés par l’ARS à conserver des embryons dans le cadre d’une activité d’aide médicale à la procréation n’ont pas besoin d’obtenir une autorisation supplémentaire de l’ABM (Article L2151-9 CSP). Plus d’informations sur cette procédure sur le site de l’ABM ici.
Les recherches visant à obtenir des cellules souches embryonnaires humaines (CSEh) à partir d’embryons doivent obtenir une autorisation de l’ABM (Article L2151-5 CSP). Celle-ci est délivrée sous quatre mois et l’absence de réponse vaut refus (Article R2151-6 CSP). Plus d’informations sur cette procédure sur le site de l’ABM ici.
Les établissements stockant des CSEh doivent faire une déclaration auprès de l’ABM (Article L2151-9 CSP). Plus d’informations sur cette procédure sur le site de l’ABM ici.
L’importation ou l’exportation de CSEh à des fins de recherche doit être préalablement autorisées par l’ABM (Article L2151-8 CSP). Le silence gardé après quatre mois vaut rejet de la demande. Les autorisations sont délivrées pour une durée de 2 ans. Plus d’informations sur cette procédure sur le site de l’ABM ici.
Toutes ces demandes sont adressées à l’ABM par courriel ([email protected]) ou par voie postale.
Échantillons biologiques humains génétiquement modifiés : différentes procédures peuvent s’appliquer en fonction du niveau de confinement et du produit considéré.
Dans le cas d’utilisations confinées d’échantillons biologiques humains génétiquement modifiés, les installations doivent être agréées par leMESR (Article L532-3 du code de l’environnement).
Lorsque cette utilisation confinée se fait dans le cadre d’une recherche clinique, une déclaration à l’ANSM est requise (Articles R532-35 et suivants du code de l’environnement) ; la mise en œuvre de l’utilisation confinée étant subordonnée à l’autorisation de la RIPH. Plus d’informations sur les déclarations d’utilisations confinées sont disponibles sur le site de l’ANSM ici.
Dans le cas des disséminations volontaires dans le cadre d’une recherche clinique, une autorisation du Ministère de l’environnement est requise (article R533-21 du code de l’environnement). Les demandes sont à réaliser via la plateforme Démarche numérique.
Pour les médicaments de thérapie innovante soumis à la procédure centralisée d’autorisation de mise sur le marché (évaluation par l’EMA et décision de la Commission européenne) et composés d’Organismes Génétiquement Modifiés (OGM), l’autorisation de mise sur le marché vaut autorisation de dissémination volontaire des OGM (Article R533-49 I. du code de l’environnement).
Dans le cadre des demandes d’autorisations d’accès précoces, d’accès compassionnel et de médicaments de thérapie innovante préparés ponctuellement de thérapie génique, l’autorisation de dissémination volontaire des OGM est délivrée par l’ANSM selon les procédures définies à l’article R533-49 II. du code de l’environnement.
Plus d’informations sur les autorisations de dissémination volontaire sont disponibles sur le site de l’ANSM ici.
Contact possible avec le ministère de l’environnement : [email protected].
Législation de l’Union européenne
Directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins, et modifiant la directive 2001/83/CE, JO L 033 du 08/02/2003 p. 0030 - 0040
Texte original (disponible dans les 24 langues officielles de l’UE)
Directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains, JO L 102 du 7.4.2004, p. 48–58
Texte original (disponible dans les 24 langues officielles de l’UE)
Règlement (UE) n ° 536/2014 du Parlement européen et du Conseil du 16 avril 2014 relatif aux essais cliniques de médicaments à usage humain et abrogeant la directive 2001/20/CE, JO L 158 du 27.5.2014, p. 1–76.
Texte original (disponible dans les 24 langues officielles de l’UE)
Règlement (UE) 2016/679 du Parlement européen et du Conseil du 27 avril 2016 relatif à la protection des personnes physiques à l'égard du traitement des données à caractère personnel et à la libre circulation de ces données, et abrogeant la directive 95/46/CE (règlement général sur la protection des données), JO L 119 du 4.5.2016, p. 1–88.
Texte original (disponible dans les 24 langues officielles de l’UE)
Règlement (UE) 2024/1938 du Parlement européen et du Conseil du 13 juin 2024 concernant les normes de qualité et de sécurité des substances d’origine humaine destinées à une application humaine et abrogeant les directives 2002/98/CE et 2004/23/CE, PE/8/2024/REV/1, JO L, 2024/1938, 17.7.2024.
Texte original (disponible dans les 24 langues officielles de l’UE)
Au niveau européen, plusieurs lignes directrices et instruments de droit souple sont pertinents pour les activités des biobanques, notamment pour la gestion des données de santé.
Avis du Groupe européen d'éthique des sciences et des nouvelles technologies (GEE) : le GEE a rendu plusieurs avis sur les questions éthiques liées aux biobanques.
Aspects éthiques des banques de tissus humains, Opinion N° 11, 21 juillet 1998
Aspects éthiques des banques de sang du cordon ombilical, Avis N° 19, 16 mars 2004
Avis sur les aspects éthiques de la recherche sur les cellules souches humaines et leur utilisation, Avis N°15, 14 novembre 2005
Lignes directrices concernant la gestion des données en biobanques.
Le Règlement Général sur la Protection des Données (RGPD) encourage la rédaction de codes de conduite pour contribuer à la bonne application du règlement et la convergence des pratiques (articles 40 et 41 du RGPD). Plusieurs initiatives européennes sont en cours.
La première est menée par le BBMRI-ERIC dont l’objectif est d’élaborer un code sectoriel complet. BBMRI-ERIC, Code of Conduct for Health Research [en cours de rédaction].
La seconde initiative est menée par GÉANT et concerne le traitement des données à caractère personnel dans le secteur de la recherche et de l'éducation. GEANT, Data protection Code of Conducts, GÉANT Code of Conduct (CoCo) for Service Providers using identity federations.
La troisième initiative est portée par l’EUCROF (European Contact Research Organizations Federation) au sujet des participants à des essais cliniques. EUCROF, Code of Conduct For Service Providers in Clinical Research, 12 septembre 2024.
Lignes directrices et avis du Comité européen de la protection des données (EDPB) et du contrôleur européen de la protection des données. (en anglais)
European Data Protection Board, Guidelines 1/2019on Codes of Conduct and Monitoring Bodies under Regulation 2016/679
European Data Protection Board, Opinion 3/2019 concerning the Questions and Answers on the interplay between the Clinical Trials Regulation (CTR) and the General Data Protection regulation (GDPR) (Art. 70.1.b))
Organoid biobanking: identifying the ethics - BOERS Sarah N, VAN DELDEN Johannes JM, CLEVERS Hans et al., EMBO reports, 17, John Wiley & Sons, Ltd, juillet 2016, no 7, p. 938‑941.
Research Biobanking, Personal Data Protection and Implementation of the GDPR in France - CHASSANG Gauthier, HISBERGUES Michael et RIAL-SEBBAG Emmanuelle, in Slokenberga Santa, Tzortzatou Olga et Reichel Jane (éd.), GDPR and Biobanking: Individual Rights, Public Interest and Research Regulation across Europe, Cham, Springer International Publishing, 2021, p. 257‑276.
Biobanking with genetics shapes precision medicine and global health - GALLAGHER C. Scott, GINSBURG Geoffrey S. et MUSICK Anjené, Nature Reviews Genetics, Nature Publishing Group, novembre 2024, p. 1‑12.
Biobanks for Europe – A challenge for governance : Report of the expert group on dealing with ethical and regulatory challenges of international Biobank Research - GOTTWEIS, Herbert., European Union, 2012, p. 20.
Responsible Research with Human Tissues: The Need for Reciprocity Toward Both Collectives and Individuals - LENSINK Michael A., JONGSMA Karin R., BOERS Sarah N. et al., The American Journal of Bioethics, 21, Taylor & Francis, avril 2021, no 4, p. 75‑78.
Bioéthique - L’existence des contraintes légales et réglementaires des biobanques -MESSAOUDI Zeineb, SOLTANI Nisrine et ARRIGHI Nicole, médecine/sciences, 36, EDP Sciences, mars 2020, no 3, p. 279‑282.
La réglementation des biobanques et des banques de données de santé en Europe : Etude de droit comparé - TALANOVA Vladislava et SPRUMONT Dominique, Institut de droit de la santé, Université de Neufchâtel, 2018.
Biobanks and the Public. Governing Biomedical Research Resources in Europe - BIOBANKING AND BIOMOLECULAR RESOURCES RESEARCH INFRASTRUCTURE (BBMRI), 2013, p. 44. Available from: BBMRI_Master_Galley.indd (bbmrieric.eu).
Evaluation of Legislation and Related Guidelines on the Procurement, Storage and Transfer of Human Tissues and Cells in the European Union – an Evidence-Based Impact Analysis -TISS.EU, 2011.
The EU General Data Protection Regulation, Answers to Frequently Asked Questions - BBMRI-ERIC, BBMRI-ERIC, 2017.
Instruments régionaux et internationaux
Conseil de l’Europe, Convention pour la protection des droits de l’homme et de la dignité de l’être humain à l’égard des applications de la biologie et de la médecine : Convention sur les droits de l’homme et la biomédecine, ouverte à la signature le 4 avril 1997 à Oviedo (Espagne), entrée en vigueur le 1er décembre 1999, STE n° 164.
Conseil de l’Europe, Comité des Ministres, Recommandation Rec(2006)4 aux États membres sur la recherche utilisant du matériel biologique d’origine humaine, adoptée le 15 mars 2006, en ligne.
Conseil de l’Europe, Comité des Ministres, Recommandation CM/Rec(2016)6 aux États membres sur la recherche utilisant du matériel biologique d’origine humaine, adoptée le 11 mai 2016, en ligne.
Protocole de Nagoya sur l’accès aux ressources génétiques et le partage juste et équitable des avantages découlant de leur utilisation, relatif à la convention sur la diversité biologique, adopté le 29 octobre 2010 à Nagoya (Japon), entré en vigueur le 12 octobre 2014, en ligne.
Association Médicale Mondiale, Déclaration d'Helsinki de l'AMM – Principes éthiques applicables à la recherche médicale impliquant des êtres humains, adoptée en juin 1964 à Helsinki (Finlande), révisée en octobre 2013, en ligne.
Association médicale mondiale, Déclaration de Taipei sur les principes éthiques relatifs à la recherche biomédicale impliquant des sujets humains dans les pays en développement, adoptée en 2002, révisée en 2016, en ligne.
Lignes directrices
Conseil des Organisations Internationales des Sciences Médicales (CIOMS), Lignes directrices internationales d’éthique pour la recherche en matière de santé impliquant des participants humains, 4e édition, Genève, 2016, en ligne.
Organisation de coopération et de développement économiques (OCDE), Les centres de ressources biologiques : Fondements du développement des sciences de la vie et des biotechnologies, Paris, OCDE, 2001.
International Society for Stem Cell Research (ISSCR), « ISSCR Guidelines for Stem Cell Research and Clinical Translation », version 1.2, août 2022, en ligne.
Normes techniques et standards
Organisation internationale de normalisation (ISO), ISO 20387:2018 – Biotechnologie – Biobanques – Exigences générales concernant la compétence des biobanques, Genève, ISO, 2018, en ligne.
ISSCR, Standards for Human Stem Cell Use in Research, juin 2023, en ligne.
ISSCR, The ISSCR Guide to Stem Cell Treatments, juin 2023, en ligne.
ANSM, Guide des bonnes pratiques relatives au prélèvement de tissus et de cellules du corps humain sur une personne vivante ou décédée, en vue d’une utilisation thérapeutique, Février 2020.
Adrien Bottacci, Doctorant en droit européen de la santé – Communication manager of the EAHL Interest Group on Supranational Biolaw - Centre de droit de la santé (CDSA) - Centre d'Etudes et de Recherches Internationales et Communautaires (CERIC) – Aix-Marseille Université, France
Aurélie Mahalatchimy, Responsable EuroGCT du Work Package 4, UMR 7318 DICE CERIC, Aix-Marseille Université, CNRS, Aix-en-Provence, France