A Epidermólise Bolhosa (EB) é uma doença genética que altera a forma como células e tecidos são estruturados. Existem diferentes tipos de EB, mas um sintoma comum à todos eles é a fragilidade extrema da pele, que facilmente se desfaz e leva à formação de bolhas e machucados a partir de fricções levels, estresse e pequenos ferimentos. Atualmente não há cura para a EB, apesar de pesquisas médicas e alguns tratamentos em fase clínica de investigação terem demonstrado resultados bastante promissórios. Como a terapia gênica e celular podem ajudar?
O que nós sabemos?
Todas as formas de EB são causadas por mutações em genes, que alteram o código que leva à formação proteínas importantes para a conexão entre células e tecidos. EB Simples, EB Juncional, EB Distrófica, e EB Kindler são os quatro tipos primários de EB.
A EB é uma doença relativamente rara, com uma incidência estimada de 1 caso a cada 50,000 nascimentos.
A EB afeta visivilmente a pele, mas essa doença também pode causar complicações em outros órgãos, como bolhas e machucados na garganta, nas vias respiratórias e no trato urinário. A EB pode progredir para uma doença sistêmica com consequências fatais, assim como pode levar ao desenvolvimento de câncer de pele agressivo.
O que os pesquisadores estão investigando?
Existem diversos novos equipamentos, medicamentos e tratamentos que estão sendo estudados em pesquisas clínicas. A maioria desses estudos tem como objetivo melhorar o manejo da EB. Alguns estudos buscam “corrigir” as mutações genéticas que estão por trás do desenvolvimento da EB. A pesquisa para desenvolvimento de terapias gênicas pode tornar possível a capacidade de restaurar genes saudáveis em células da pele.
Trabalhar com células-tronco da pele, otimizar como cultivar a pele em laboratórios e melhorar os métodos de transplante de tecido são componentes chave para desenvolver terapias gênicas com sucesso contra a EB.
Quais são os desafios?
Tratar uma doença genética é bastante desafiador visto que a causa da doença está no DNA da pessoa.Tratamentos para compensar as mutações genéticas devem reparar o gene mutante ou adicionar uma cópia funcional do gene nas células. Isto não é uma tarefa simples. Modificar o DNA celular ou adicionar novos genes nas células apresenta possíveis riscos, como a possibilidade de causar câncer. Os transplantes de célula e de tecido apresentam riscos como sangramento, infecção e rejeição do transplante.
Um outro desafio a ser superado é fazer com que tratamentos complexos, como as terapias gênicas, sejam acessíveis para todos.
Sobre a Epidermólise Bolhosa
Epidermólise Bolhosa (EB) se refere à um grupo de doenças genéticas raras do tecido conjuntivo que resulta numa pele extremamente frágil e com muitas bolhas. As bolhas podem ser causadas pela fricção com a pele, pequenos machucados na pele e durante atividades diárias, como se coçar ou esfregar a pele. Essas bolhas podem se tornar machucados muito doloridos e causar feridas abertas. Em diversos tipos de EB, bolhas e machucados também podem se desenvolver em outros tecidos, como na boca, garganta, estômago, trato respiratório superior e trato urinário.
A gravidade dos diferentes tipos de EB é bastante abrangente. Em casos moderados, é necessário o manejo de pequenas bolhas durante toda a vida, enquanto que em outros casos, observa-se os sintomas de EB grave durante a infância, mas que podem melhorar com a idade. Formas alternativas de EB podem apresentar risco à vida visto que uma grande quantidade de pele é perdida. Outros tipos podem causar cicatrização extensa, a fusão de dedos da mão e do pé, assim como outras complicações que apresentam risco de vida. Algumas formas da doença podem levar ao desenvolvimento do câncer de pele, reduzindo a expectativa de vida.
Como a EB é rara, estimar o número total de pessoas que tem EB é desafiador. A DEBRA, organização beneficente dedicada à EB, estima que aproximadamente 500,000 pessoas ao redor do mundo apresentem algum tipo de EB.
Todos os tipos de EB que são hereditários são causados por mutações no DNA. Essas mutações alteram um dos diversos genes importantes na formação de proteínas que conectam as células e os tecidos de forma conjunta. Por isso que a EB também é conhecida como uma doença do “tecido conjuntivo”. Atualmente, existem 16 genes no nosso DNA que foram associados à EB hereditária clássica.
Uma mutação em um gene ligado à EB é passado dos pais para a criança. Nosso DNA contém duas cópias de cada gene (exceto os genes presentes no cromossomo X e Y). Em diversas formas de EB, é necessário herdar somente uma cópia de um gene com mutação ligada à EB. Isto é conhecido como a forma “dominante” da EB. O progenitor que passa a forma dominante do gene com mutação ligada à EB também tem EB e sintomas associados. Outras formas de EB requerem o gene com mutação ligada à EB de ambos os pais. Essas são conhecidas como as formas “recessivas” da EB. As formas recessivas da EB são geralmente inesperadas para a família porque nenhum dos pais apresenta sintomas de EB e não sabem que carregam a mutação.
Sobre a pele
Nossa pele é um órgão extenso e complexo. Esse órgão age como um escudo de proteção à água, mantendo a água e bactérias do lado de fora. Ela nos protege do vento e do clima, e também nos ajuda a regular a temperatura do corpo. É na pele que a vitamina D é sintetizada. Além disso tudo, nossa pele possui diversas células especializadas que nos permite sentir a temperatura, textura, pressão e dor. Nossa pele é feita para ser resistente, isto é, se tudo sai conforme o planejado.
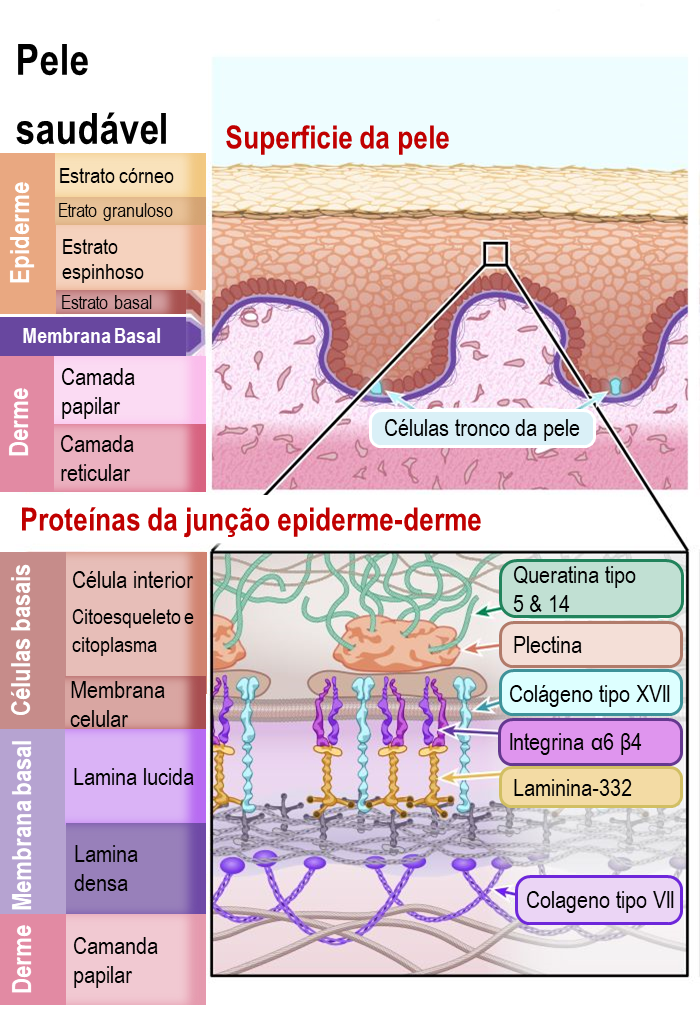
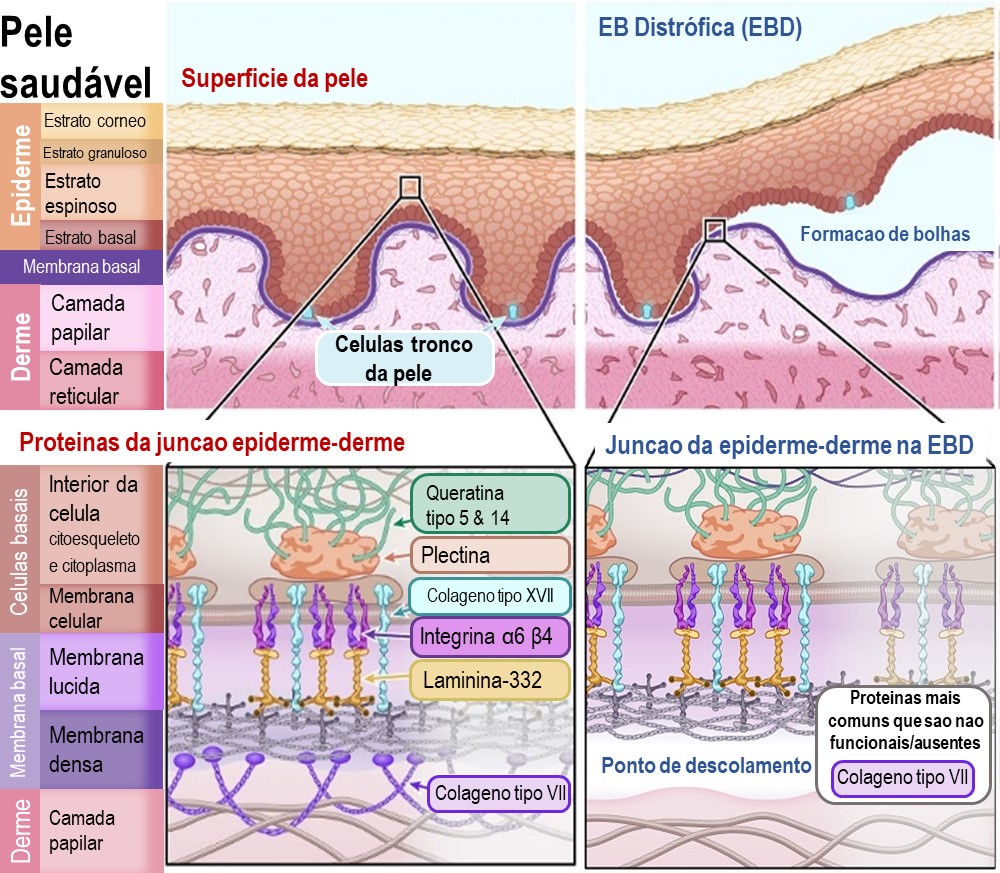
As duas principais camadas da pele são a epiderme (a camada mais externa) e a derme. Os pesquisadores ainda subdividem essas camadas em camadas mais específicas (ou estratos) de células. Você pode observar essas sub-camadas no diagrama.
A epiderme é composta majoritariamente por queratinócitos. Eles são denominados queratinócitos pelo fato de produzirem uma proteína bem forte chamada queratina – que é a mesma proteína que forma nossos fios de cabelo e unhas. Os queratinócitos se desenvolvem a partir de células tronco da pele que residem no estrato basal, uma sub-camada da epiderme, e nos folículos pilosos da derme. As células tronco da pele estão continuamente produzindo novos queratinócitos para substituir as células da pele que são continuamente perdidas. De fato, quase todas as células da camada mais externa da pele são substituídas a cada 4 a 5 semanas! Isso faz com que as células tronco da pele sejam muito importantes para a manutenção da saúde da pele.
A derme é uma camada da pele que contem os vasos sanguíneos, células sensoriais de tato, temperatura e dor, as glândulas sebáceas, folículos pilosos, entre outros. Existem diferentes tipos de células na derme, mas a maior parte da derme é composta por uma rede de fibras conectivas e proteínas. Essa rede de proteínas promove o suporte da estrutura da pele e mantém tudo no lugar. As proteínas envolvidas nessa rede são principalmente o colágeno e a elastina.
O ponto de conexão entra a epiderme e a derme também é importante, principalmente quando se trate da epidermólise bolhosa. O ponto de junção é chamado de “membrana basal” e é uma camada fina de tecido conjuntivo importante para a ligação entre a epiderme e a derme. A membrana basal é primariamente composta de dois tipos de proteína, a laminina e o colágeno.
Apesar de todas as formas de EB gerarem bolhas como um sintoma, elas não são causadas pelo mesmo problema. O gene afetado por uma mutação que está ligada à EB determina aonde, por que e como ocorre a extensa formação de bolhas e a desintegração da pele. A mutação também determina qual tipo de EB uma pessoa tem.
Atualmente, existem 16 genes específicos no nosso DNA que foram ligados à EB clássica. Mutações nesses genes previnem que as células produzam proteínas funcionais que mantém a epiderme e a derme juntas. Saber o papel que as proteínas normalmente possuem na manutenção da estrutura da pele é essencial para entender como a EBS, a EBJ e a EBD são diferentes. Elas também revelam porque alguns tipos de EB são mais severos que outros e porque um tratamento para certo tipo de EB pode não funcionar para os outros.
Abaixo encontra-se um resumo que ressalta os genes, as proteínas e algumas especificidades sobre aonde e como o descolamento ocorre para os tres principais tipos de EB.
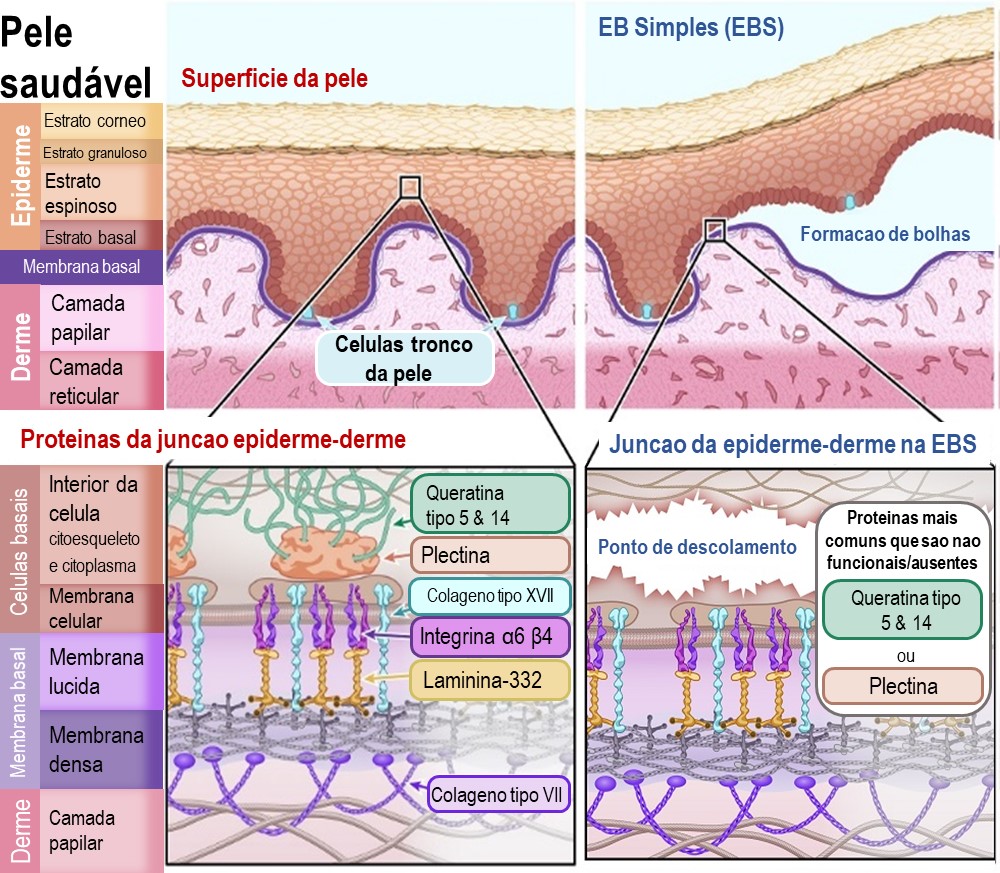
Epidermólise Bulhosa Simples (EBS):
Na EBA, a separacao da epiderme com a derme ocorre na camada de celulas basais da epiderme (Estrato basal), logo acima da membrana basal. O problema e que as proteinas que seguram as celulas basais na membrana basal nao se aderem propriamente ao suporte estrutural dentro das celulas (o citoesqueleto). Isso resulta no rasgo e na ruptura das celulas basais, deixando a parte debaixo dessas celulas aderidas a membrana basal.
Os tipos primarios de proteinas afetados por mutacoes genicas na EBS sao:
Queratina tipo 5 - [Nome do gene: KRT5]
Queratina tipo 4 - [Nome do gene: KRT14]
Plectina - [Nome do gene: PLEC1]
Ilustração 2: Diagrama de EB Simples
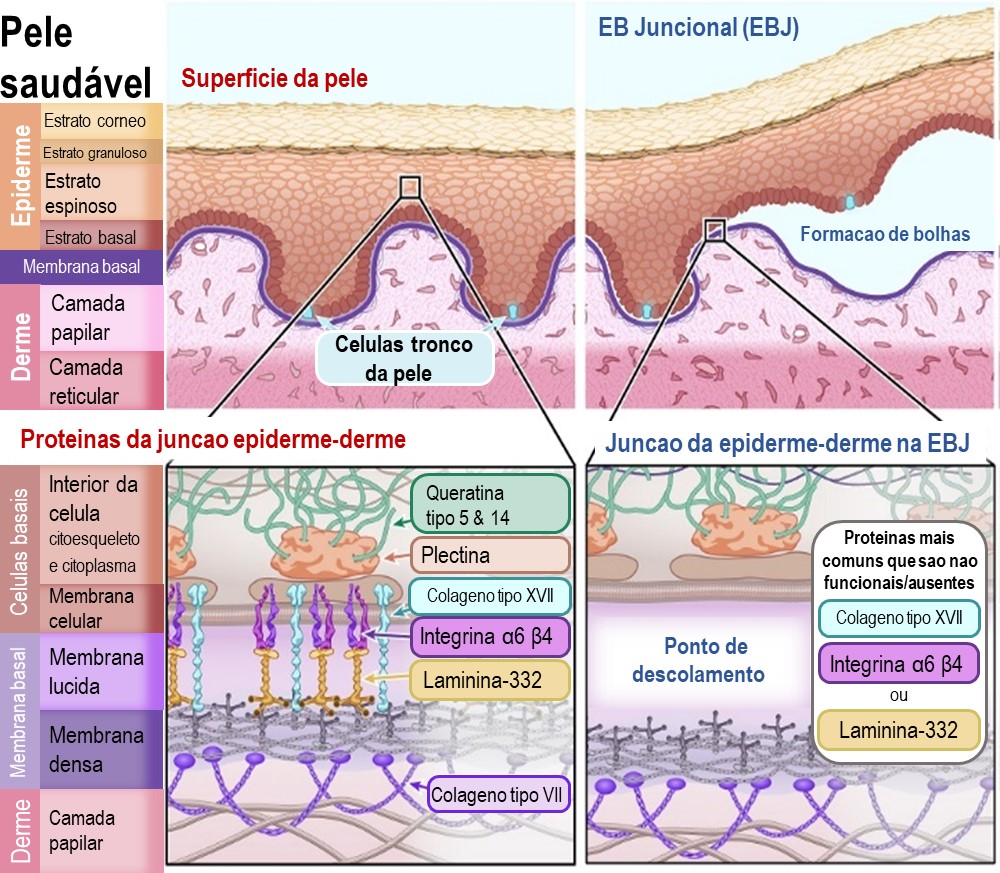
Epidermólise Bulhosa Juncional (EBJ)
Na EBJ a separação da epiderme e da derme ocorre na membrana basal, especificamente na subcamada da “lamina lucida”. Esta subcamada é composta por proteínas responsáveis pela ancoragem das células basais da epiderme à derme. Quando as proteínas desta camada estão ausentes ou não funcionais, este local de junção da membrana basal não se mantém intacto.
As principais proteínas afectadas por mutações genéticas na EBJ são:
Colagénio tipo XVII (também conhecido por BPAG2) – [nome do gene: COL17A1]
Integrina a6b4 (subunidades alfa 6, beta 4) – [nome dos genes: ITGA6 e ITGB4]
Laminina-332 (também conhecida por laminina-5) – [nome dos genes: LAMA3, LAMB3 e LAMC2]
Ilustração 3: Diagrama da EB Juncional
Epidermólise Bulhosa Distrófica (EBD)
Na EBD a separação da epiderme e da derme ocorre abaixo da membrana basal. A principal proteína que liga a membrana basal à derme é o colagénio VII. Esta proteína forma fibrilhas de ancoragem que se enlaçam nas proteínas estruturais da derme. Sem colagénio VII, a membrana basal separa-se facilmente da derme.
A principal proteína afectada por uma mutação genética em EBD é:
Colagénio tipo VII – [Nome do gene: COL7A1]
Ilustração 4: Diagrama de EB Distrófica
Epidermólise Bulhosa Kindler (EBK)
A EB Kindler (EBK) pode afectar múltiplas e diferentes camadas da pele. Quando a proteína afectada esta ausente ou disfuncional, perturba a capacidade de crescimento e divisão dos queratinócitos (células da pele), assim como a capacidade de estabelecer a adesão da epiderme à derme.
A principal proteína afectada por uma mutação genética na EBK é:
Kindlina-1- [Nome do gene : FERMT1]
Nossos parceiros na Rede de Pesquisa EB tem informações sobre a EB e suas classificações
Actualmente, não existem terapias causais aprovadas para EB que corrijam as mutações genéticas associadas subjacentes. Os tratamentos focam-se principalmente em prevenir ou aliviar os sintomas de EB. Medidas preventivas podem envolver o uso de ligaduras ou pensos almofadados para minimizar a fricção, atrito e minimizar o traumatismo da pele. Os tratamentos podem também prevenir infeções e promover a cicatrização de bolhas e feridas.
Em casos mais graves de EB, medicamentos podem ser utilizados para prevenir infeções de feridas abertas. Uma cirurgia pode também ser realizada para prevenir fusão de dedos dos pés ou das mãos, ou o estreitamento da garganta e do esófago.
Em casos extremos de EB, os médicos podem cobrir grandes feridas abertas com enxertos de pele. No entanto, o enxerto de pele de dadores é muito raro devido à elevada taxa de rejeição pelo sistema imunitário, mesmo se o dador e o paciente forem compatíveis. Deste modo, os médicos utilizam frequentemente substitutos de pele temporários para cobrir grandes feridas abertas. Novos ‘substitutos de pele biológicos’ estão também a tornar-se cada vez mais comuns.
Investigação actual
Melhorar a qualidade de vida
Actualmente não existem tratamentos clinicamente aprovados que resolvam as causas biológicas subjacentes à EB. No entanto, existem diversos investigadores, empresas farmacêuticas e de produtos médicos a trabalhar para desenvolver produtos para melhorar a gestão dos sintomas de EB. Os produtos que estão a ser investigados vão desde melhores sistemas de ligaduras e pensos a pomadas e loções que ajudem a pele a sarar. Actualmente estão a decorrer vários ensaios clínicos para testar diferentes tipos de pensos para feridas e medicamentos em pomada com o objectivo de acelerar o processo de cicatrização da pele em indivíduos com EB.
Em casos severos de EB, os médicos podem necessitar de cobrir feridas abertas de grandes dimensões. Frequentemente isto envolve cobrir feridas com substitutos de pele sintética. No entanto, há empresas que lançaram recentemente ‘substitutos de pele biológicos’ mais avançados, como o Biobrane®, Integra®, Orcel®, Apligraft® e outros. Estes substitutos de pele avançados tem como objectivo melhorar a cicatrização usando matrizes sintéticas em que as células gostam de crescer, proteínas que se encontram na pele e por vezes em queratinócitos (células da pele) vivos. Os exemplos anteriores são apenas algumas das novas tecnologias e produtos que estão a ser desenvolvidos e que podem ajudar a melhorar significativamente a qualidade de vida daqueles que sofrem de EB.
Produzir pele ‘nova’
A forma ideal de curar uma doença genética é corrigir ou substituir o gene mutado no DNA do paciente. Os investigadores estão a criar terapias genéticas que fazemisto mesmo. Vários ensaios clínicos e tratamentos tem sido bem sucedidos em anos recentes. Estes resultados encorajadores indicam que os tratamentos estão próximos. Pela primeira vez pode estar para breve tratamentos que resolvem os problemas que causam EB.
As terapias genéticas estão actualmente nas fases iniciais de desenvolvimento. Novas descobertas e tecnologias, como edição genética por CRISPR-Cas9, estão a acelerar a forma como o DNA pode ser editado em células. No entanto, alterar o DNA nas células de uma pessoa não é uma tarefa fácil. O processo pode também envolver riscos elevados. Se a adição ou edição de um gene correr mal pode causar cancro, adicionar novas complicações e piorar o estado de um indivíduo. Os ensaios clínicos são importantes para identificar riscos associados a novos tratamentos e demonstrar que estes funcionam.
Em muitos casos, as células estaminais acompanham as terapias genéticas. Em terapias genéticas de EB, os investigadores tem como objectivo editar o DNA de células estaminais da pele e não de outros tipos de células da pele. Isto porque adicionar (ou editar) um gene em células estaminais da pele levará a que esse gene seja transmitido a todas as restantes células da epiderme produzidas pelas células estaminais. Isto é importante quando quase todos os queratinócitos na nossa pele são substituídos a cada 4-5 semanas. Adicionalmente, as células estaminais da pele estão em renovação contínua, o que significa que quaisquer alterações ao DNA introduzidas numa célula estaminal tem o potencial de se tornar ‘permanentes’ (ou durar pelo menos tanto tempo como as células estaminais). Vários tratamentos para EB que estão actualmente em ensaios clínicos estão a testar métodos de editar o DNA de células estaminais da pele do paciente para criar pele ‘nova’ saudável para transplante. Apesar de os métodos e detalhes destes diferentes ensaios variarem, a ideia geral e o processo são muito semelhantes. Envolve retirar amostras de pele de uma pessoa que sofre de EB e cultivar estas células em laboratório, na expectativa de que as amostrar contenham células estaminais da pele. Métodos de edição de DNA são depois utilizados para adicionar ou editar genes nas células e restaurar a produção normal de proteína. Todas as células estaminais da pele que contenham as novas edições no DNA são posteriormente utilizadas em métodos avançados de cultura para gerar camadas de pele em laboratório. Após este processo, as células podem ser transplantadas no paciente. Algumas destas abordagens envolvem o uso de células estaminais pluripotentes induzidas (iPSC), um tipo de célula estaminal que pode ser gerado a partir das células do nosso corpo. Estas células podem ser propagadas indefinidamente e dar origem a qualquer tipo de célula da pele. Elas podem representar uma fonte única de células para substituir tecido danificado. Apesar de as células estaminais pluripotentes induzidas (iPSCs) terem sido aprovadas em ensaios clínicos para várias doenças, pacientes que receberam transplantes de células derivadas de iPSC podem ter um risco acrescido de desenvolver cancro.
Este tipo de tratamento requer muito tempo e trabalho para identificar o problema genético para cada pessoa afectada por EB, desenvolver um processo específico de edição de DNA, semanas para crescer células e cirurgiões clínicos para executar os transplantes de pele. Requer também infraestruturas especializadas para a investigação e expansão de células. Apesar destes obstáculos, têm sido reportados grandes avanços que demonstram que este processo pode resultar. A próxima secção discute um dos casos de sucesso.
Foco na investigação europeia: Um caso de sucesso
Em 2017, um grupo de investigadores e médicos liderado pelo Professor Michele De Luca divulgou o sucesso da utilização de terapia genética e enxertos de pele criados em laboratório para salvar um rapaz que tinha perdido 80% da pele. O rapaz sofria de EBJ provocada por mutações no gene LAMB3, herdado de ambos os progenitores. Todos os tratamentos, incluindo diversas abordagens extremas, tinham falhado e os médicos pensavam que o rapaz tinha uma baixa probabilidade de sobreviver. As autoridades governamentais autorizaram o ‘uso compassivo’ de um tratamento genético preliminar que só tinha sido testado previamente em dois estudos de caso em que apenas um paciente foi observado, respectivamente. Os pais do rapaz também concordaram com o procedimento, apesar de terem sido informados de que o rapaz poderia não sobreviver ao procedimento em si.
Os investigadores começaram por recolher amostras de pele do rapaz e expandir estas células em laboratório. Esperavam obter agregados de células que continham células estaminais da pele. Os agregados de células foram subsequentemente tratados com um vírus para adicionar uma copia funcional do gene LAMB3 ao DNA das células estaminais da pele. Este processo não resolve a mutação que o rapaz tinha nas suas próprias cópias do gene. Em vez disso, novo DNA foi adicionado com uma cópia do gene LAMB3 as células estaminais da pele do rapaz. Este novo gene permite que as células produzam proteínas de laminina funcionais. As células estaminais da pele com o gene funcional LAMB3 que foi adicionado foram utilizadas para crescer camadas de pele em laboratório utilizando técnicas de cultura de células avançadas. Este processo demora várias semanas. As pequenas peças de pele criada em laboratório foram transplantadas no rapaz em dois procedimentos distintos com um mês de intervalo. No final, o rapaz sobreviveu e continua a viver com pele criada a partir de células em que o DNA foi modificado em mais de 80% do corpo. Os investigadores examinaram esta pele e constataram que parece extremamente semelhante à pele normal em diversos aspectos. O sucesso deste tratamento é muito entusiasmante; há até quem diga que o resultado foi milagroso porque excedeu as expectativas. Infelizmente, tratamentos de terapia genética como estes não estão ainda aprovados clinicamente ou universalmente acessíveis. É também importante compreender que terapias genéticas deste tipo devem ‘corrigir’ problemas causados por genes mutados específicos.
Medicina personalizada como esta é dispendiosa, requer infraestruturas especiais e o trabalho de muitas pessoas. Espera-se que tratamentos como este sejam aprovados clinicamente, acessíveis e disponíveis para diversas formas de EB.
O futuro é promissor. O sucesso do caso descrito anteriormente, tal como os resultados positivos de outros estudos, serão origem a vários novos ensaios clínicos. M. De Luca do Centro de Medicina Regenerativa ‘Stefano Ferrari’ em Modena, Itália, em conjunto com JW Bauer do Hospital Universitário de Dermatologia em Salzburgo, Áustria, lideram vários ensaios clínicos utilizando procedimentos semelhantes mas mais avançados para substituir mutações nos genes COL7A1 e COL17A1 (Estudo 1, Estudo 2). Investigadores de outras instituições estão também a desenvolver as suas próprias terapias genéticas, como o ensaio clínico liderado por Jean Yuh Tang da Faculdade de Medicina da Universidade de Stanford, que tem também como alvo mutações no gene COL7A1 responsáveis por EBD.
Espera-se que a investigação dê origem a tratamentos para EB seguros, eficazes e acessíveis.
Outras abordagens terapêuticas
Como descrito na secção anterior, produzir pele com DNA editado tem grandes desafios; o processo é complexo, caro e arriscado. Adicionalmente, pode demorar anos até que este procedimento se torne um tratamento realista.
Estes desafios levaram os investigadores a pensar em procedimentos alternativos para o tratamento de EB que podem ser desenvolvidos mais rapidamente, clinicamente aprovados e economicamente acessíveis aqueles que são afectados por EB. Estes envolvem células estaminais, medicamentos, extratos de plantas e muitos outros tratamentos. Os dois descritos abaixo são apenas alguns exemplos.
Cremes tópicos e loções terapêuticas estão a ser desenvolvidos para minimizar os problemas originados por mutações específicas de EB. Dois estudos clínicos com particular interesse estão a utilizar cremes que tentam reparar ou substituir o gene COL7A1, o que poderia ajudar indivíduos com EBD. Um dos cremes contém componentes que permitem às células da pele ultrapassar a mutação no gene COL7A1 quando produzem a proteína do colagénio VII. O segundo creme contém partículas virais que inserem uma cópia saudável do gene COL7A1 nas células. (Estas partículas virais não conseguem se replicar, o que as torna seguras para serem utilizadas).
Os transplantes de medula óssea estão tradicionalmente associados a leucemia e outras doenças do sangue e do sistema imunitário. No entanto, alguns grupos de investigação estão a explorar o tratamento de EB com alotransplantes de medula óssea, em que as células estaminais são originárias de doadores saudáveis. A vantagem deste método é a de que os transplantes de medula óssea têm sido utilizados com sucesso há muitos anos e a técnica tem vindo a ser melhorada ao longo do tempo. Resultados preliminares sugerem que células estaminais saudáveis da medula óssea (ou células produzidas por estas células estaminais) produzem proteínas do tecido conjuntivo que estão ausentes na pele, como o colagénio VII em indivíduos com EBD. No entanto, esta abordagem tem também as suas limitações incluindo a necessidade de encontrar um doador imuno-compatível, e actualmente os riscos associados são elevados.
Uma lista mais complete de ensaios clínicos para EB pode ser encontrada em clinicaltrials.gov. (Note-se que este endereço público lista apenas ensaios clínicos. Não faz uma verificação da segurança, validade científica ou reputação das entidades que conduzem os ensaios. Por favor leia qualquer isenção de responsabilidade, fale com prestadores de cuidados de saúde e informe-se sobre riscos e potenciais benefícios).