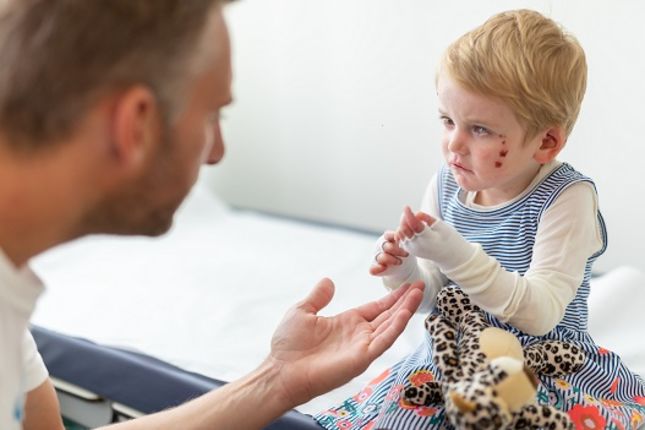
Epidermolysis Bullosa (EB) ist eine genetische Krankheit, bei welcher der Zusammenhalt von Zellen und Geweben gestört ist. Es gibt verschiedene Formen von EB, aber ein Symptom was bei allen Formen von EB auftritt, ist extrem zerbrechliche Haut, die selbst bei wenigReibung oder kleinen Verletzungen leicht reißt oder Wunden und Blasen bildet. Derzeit gibt es keine Heilung für EB, aber die Gentherapie wird bereits zur Behandlung von Menschen mit EB eingesetzt, und vielversprechende Forschungsarbeiten und klinische Studien untersuchen neue Möglichkeiten, wie Gen- und Zelltherapie helfen können.
Was wissen wir?
Viele neue Geräte, Medikamente und biologische Behandlungen werden in klinischen Studien untersucht. Viele dieser Studien versuchen die Symptome von EB zu verbessern. Einige Studien zielen darauf ab, die genetischen Mutationen zu „reparieren“, die EB verursachen.
Die Entwicklung von Gentherapien könnte es ermöglichen gesunde Gene in kranken Hautzellen wiederherzustellen. Schlüsselkomponenten für die Entwicklung erfolgreicher Gentherapien für EB sind das Arbeiten mit Hautstammzellen, die Optimierung des Wachstums dieser Zellen im Labor und die Verbesserung von Gewebetransplantationsmethoden.
Woran arbeitet die Forschung?
Viele neue Geräte, Medikamente und biologische Behandlungen werden in klinischen Studien untersucht. Viele dieser Studien versuchen die Symptome von EB zu verbessern. Einige Studien zielen darauf ab, die genetischen Mutationen zu "reparieren", die EB verursachen.
Die Entwicklung von Gentherapien könnte es ermöglichen gesunde Gene in kranken Hautzellen wiederherzustellen. Schlüsselkomponenten für die Entwicklung erfolgreicher Gentherapien für EB sind das Arbeiten mit Hautstammzellen, die Optimierung des Wachstums dieser Zellen im Labor und die Verbesserung von Gewebetransplantationsmethoden.
Welche Herausforderungen gibt es noch?
Die Behandlung einer genetischen Krankheit ist eine große Herausforderung, da die Ursache in der DNA der betroffenen Person liegt. Um eine Genmutation zu behandeln, müsste das mutierte Gen repariert oder den Zellen eine neue funktionelle Kopie des Gens hinzugefügt werden. Dies ist keine einfache Aufgabe. Das Verändern von DNA oder das Hinzufügen neuer Gene in Zellen birgt potenzielle Risiken, wie beispielsweise die Möglichkeit, Krebs zu verursachen. Auch Zell- und Hauttransplantationen können Risiken wie Blutungen, Infektionen und Abstoßung des Transplantats bergen.
Sorgfältige klinische Forschung soll sicherstellen, dass neue Behandlungen für EB sicher und wirksam sind.
Eine weitere Herausforderung ist, komplexe Behandlungen wie Gentherapien für jedermann zugänglich und erschwinglich zu machen.
Über Epidermolysis Bullosa
Epidermolysis bullosa (EB) ist eine Gruppe seltener genetischer Erkrankungen, die zu sehr empfindlicher, blasenbildender Haut führen. Die Blasen können durch Reibungen auf der Haut, kleinere Hautverletzungen und alltägliche Aktivitäten wie Scheuern und Kratzen verursacht werden. Diese Blasen können sich zu schmerzhaften und offenen Wunden entwickeln. Bei schweren Formen von EB findet man Blasen und wunde Stellen auch in anderen Geweben, wie z. B. im Mund, Rachen, Magen, den oberen Atemwegen und den Harnwegen.
Es gibt ein breites Spektrum an Schweregraden von EB. Leichte Ausprägungen von EB können dazu führen, dass Sie lebenslang mit kleinen Blasen zu kämpfen haben. Andere haben nur als Kind schwere Symptome, die sich mit zunehmendem Alter verbessern. Andere Formen von EB können wegen des Verlusts großer Hautmengen lebensbedrohlich sein. Wieder andere Formen können die Bildung von Narben, das Zusammenwachsen von Fingern und Zehen sowie andere lebenseinschränkende Komplikationen verursachen. Einige Formen von EB können zur Entwicklung von Hautkrebs führen und damit die Lebenserwartung verringern.
EB ist selten, was Schätzungen für die Gesamtzahl der Patienten schwierig macht. DEBRA, eine Wohltätigkeitsorganisation, die sich EB widmet, schätzt, dass etwa 500.000 Menschen weltweit irgendeine Form von EB haben.
Alle erblich-bedingten Formen von EB werden durch Mutationen in der DNA einer Person verursacht. Diese Mutationen stören eines der Gene, die wichtig für Proteine sind, die Zellen und Gewebe miteinander verbinden. Deshalb wird EB auch als „Bindegewebsstörung“ bezeichnet. Mit der klassischen vererbten EB werden derzeit 16 Gene in unserer DNA in Verbindung gebracht.
Wie wird EB vererbt?
Eine mit EB verbundene Genmutation wird oft von den Eltern an die Kinder weitergegeben. Unsere DNA enthält zwei Kopien jedes Gens (mit Ausnahme der Gene auf den X- und Y-Chromosomen).Oftmals ist die Weitergabe einer Kopie einer mit EB verbundenen Genmutation ausreichend um die Krankheit zu vererben.. Dies wird als „dominante“ Form von EB bezeichnet. Der Elternteil, der eine dominante Form der EB verbundenen Genmutation weitergibt, hat selbst auch EB und die entsprechenden Symptome. Andere Formen von EB erfordern die Vererbung einer EB-assoziierten Genmutation von beiden Elternteilen. Diese werden als „rezessive“ Formen von EB bezeichnet. Rezessive Formen von EB kommen oft unerwartet für die Familie, da kein Elternteil EB Symptome hat und möglicherweise nicht weiß, dass er oder sie die Mutation trägt.
Wie wird EB vererbt? Eine mit EB verbundene Genmutation wird oft von den Eltern an die Kinder weitergegeben. Unsere DNA enthält zwei Kopien jedes Gens (mit Ausnahme der Gene auf den X- und Y-Chromosomen).Oftmals ist die Weitergabe einer Kopie einer mit EB verbundenen Genmutation ausreichend um die Krankheit zu vererben.. Dies wird als „dominante“ Form von EB bezeichnet. Der Elternteil, der eine dominante Form der EB verbundenen Genmutation weitergibt, hat selbst auch EB und die entsprechenden Symptome. Andere Formen von EB erfordern die Vererbung einer EB-assoziierten Genmutation von beiden Elternteilen. Diese werden als „rezessive“ Formen von EB bezeichnet. Rezessive Formen von EB kommen oft unerwartet für die Familie, da kein Elternteil EB Symptome hat und möglicherweise nicht weiß, dass er oder sie die Mutation trägt
Über die Haut
Unsere Haut ist ein großes und komplexes Organ. Sie fungiert als strapazierfähiger, flexibler und, wasserdichter Schutzschild, der Wasser im Körper hält und Bakterien fernhält. Sie schützt uns vor Wind und Wetter und hilft, unsere Körpertemperatur zu regulieren. In unserer Haut wird Vitamin D synthetisiert. Darüber hinaus verfügt sie über eine große Anzahl spezialisierter Zellen, die es uns ermöglichen, Temperatur, Texturen, Druck und Schmerz zu fühlen. Unsere Haut ist auf Langlebigkeit ausgelegt, zumindest, wenn alles nach Plan läuft.
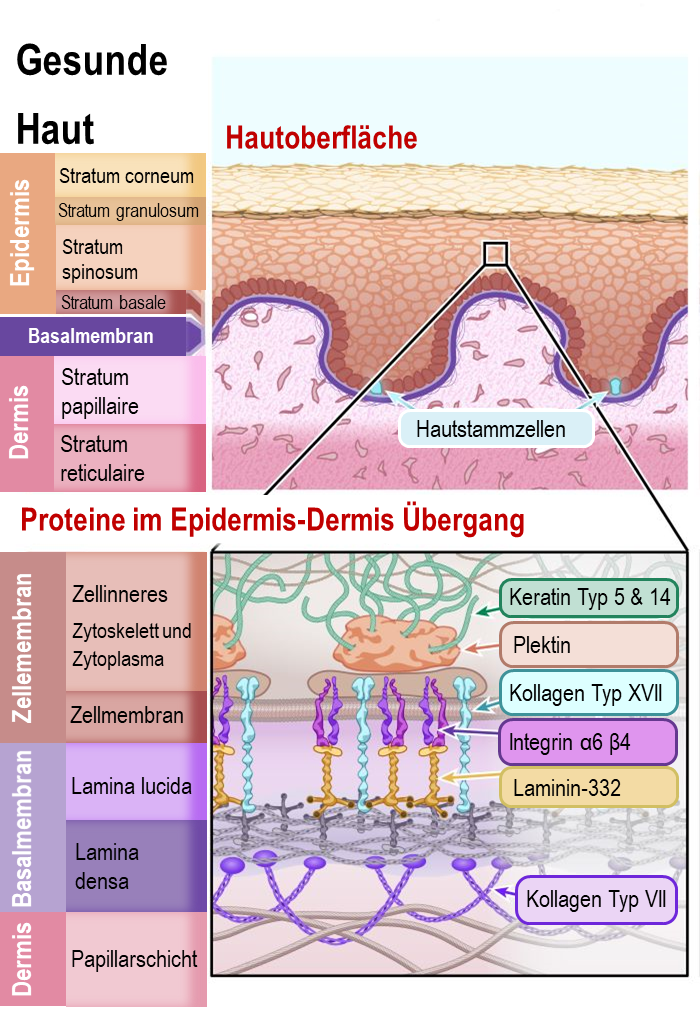
Die zwei Hauptschichten der Haut sind die Epidermis (die äußerste Schicht, auch Oberhaut) und die Dermis (auch Lederhaut). Forscher unterteilen diese Schichten weiter in spezifische Schichten von Zellen. Sie können diese Unterteilung im Diagramm sehen.
Die Epidermis besteht hauptsächlich aus Zellen namens Keratinozyten. Diese Zellen heißen so weil sie ein hartes Protein namens Keratin produzieren – dasselbe Protein, aus dem unsere Haare und Fingernägel bestehen. Keratinozyten werden aus Hautstammzellen hergestellt, die sich in der Basalschicht (Stratum Basale) der Epidermis und in den Haarfollikeln der Dermis befinden. Hautstammzellen produzieren kontinuierlich neue Keratinozyten, um die Hautzellen zu ersetzen, die absterben. Tatsächlich werden fast alle Zellen der äußeren Hautschicht alle 4 bis 5 Wochen ersetzt! Hautstammzellen sind daher sehr wichtig für die Gesundheit unserer Haut.
Die Dermis ist die Hautschicht, die Blutgefäße, Sinneszellen für Berührung, Temperatur und Schmerz, Schweißdrüsen, Haarfollikel und mehr enthält. Es gibt viele verschiedene Arten von Zellen in der Dermis aber der größte Teil besteht aus einem flexiblen Geflecht von Bindefasern und Proteinen. Dieses Proteingeflecht unterstützt die Struktur der Haut und hält alles an Ort und Stelle. Die Proteine die an diesem Netz beteiligt sind, sind hauptsächlich Kollagen und Elastin.
Auch die Verbindung zwischen Epidermis und Dermis ist wichtig, insbesondere im Zusammenhang mit EB. Diese Verbindung wird als „Basalmembran“ bezeichnet. Sie ist eine dünne Bindegewebsschicht, die dafür zuständig ist, um Epidermis und Dermis miteinander zu verbinden. Die Basalmembran besteht hauptsächlich aus zwei Arten von Proteinen, Laminin und Kollagen.
Es gibt vier Haupttypen von EB: EB simplex (EBS), junktionale EB (JEB), dystrophe EB (DEB) und Kindler-Syndrom (KEB) Diese Kategorien werden in der Regel anhand der Hautschicht unterschieden, in der sich Blasen bilden. Obwohl alle Formen von EB Blasen als Symptom haben, werden sie nicht durch das gleiche Problem verursacht. Das Gen, welches von einer EB-assoziierten genetischen Mutation betroffene ist, bestimmt wo, warum und wie stark Blasenbildung und Hautablösung auftritt. Es bestimmt auch, welche Art von EB eine Person hat.
Derzeit gibt es 16 spezifische Gene in unserer DNA, die mit klassischer EB in Verbindung gebracht werden. Mutationen in diesen Genen verhindern, dass Zellen funktionelle Proteine herstellen, die Epidermis und Dermis zusammenhalten. Das Wissen um die normale Funktion dieser Proteine für den Zusammenhalt der Haut ist entscheidend für das Verständnis der verschiedenen Arten von EB. Es erklärt auch, warum einige Arten von EB schwerwiegender sind als andere und warum die Behandlung für eine Art von EB möglicherweise nicht für alle funktioniert.
Nachfolgend sind die Gene, Proteine und einige der Details wo und warum Hautablösungen stattfinden für die drei häufigsten Formen der EB zusammengefasst.
EB Simplex (EBS)
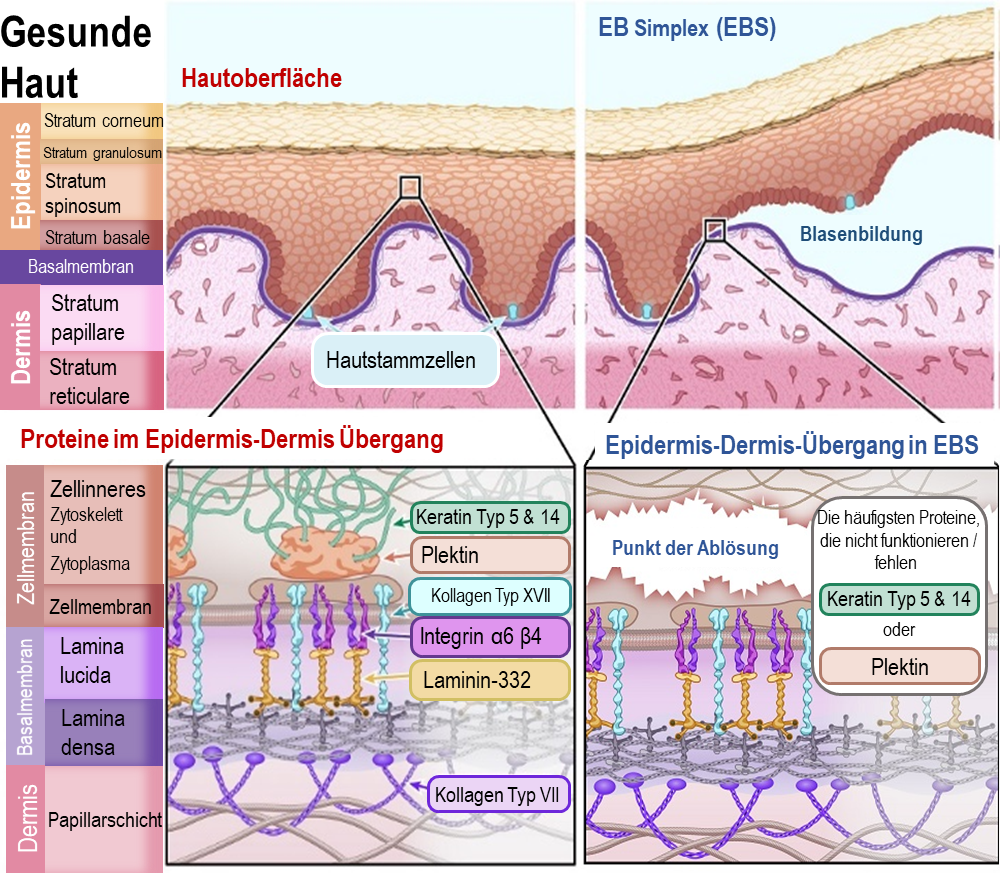
EB Simplex ist die häufigste Form von EB und macht etwa 70 % aller EB-Fälle aus. Die Schwere der EBS-Symptome kann variieren und reicht von Hautempfindlichkeit und leichten Blasen an Händen und Füßen über Fälle, in denen Blasen am ganzen Körper auftreten, bis hin zu schwereren Unterformen. EBS wird überwiegend als dominantes Merkmal von den Eltern vererbt, was bedeutet, dass nur ein Elternteil die mit EB verbundene Genmutation weitergeben muss. Dieser Elternteil hat EBS und wahrscheinlich auch die damit verbundenen Symptome (oder hatte sie).
Bei EB Simplex trennen sich Epidermis und Dermis innerhalb der Basalzellen in der Epidermis (Stratum basale), direkt über der Basalmembran. Die Proteine, die die Basalzellen an der Basalmembran befestigen, heften sich nicht richtig an die strukturelle Stütze innerhalb der Zelle (das Zytoskelett) an. Dies führt dazu, dass die Basalzellen reißen und aufbrechen, wodurch der untere Teil dieser Zellen an der Basalmembran haftet.
Die Proteine, die bei EBS am häufigsten von Genmutationen betroffen sind, sind:
Keratin Typ 5 – [Genname: KRT5]
Keratin Typ 14 – [Genname: KRT14]
Plektin – [Genname: PLEC1]
Illustration 2: Diagramm der gesunde Haut und mit Epidermolysis BullosaSimplex (EBS)
Junktionale EB
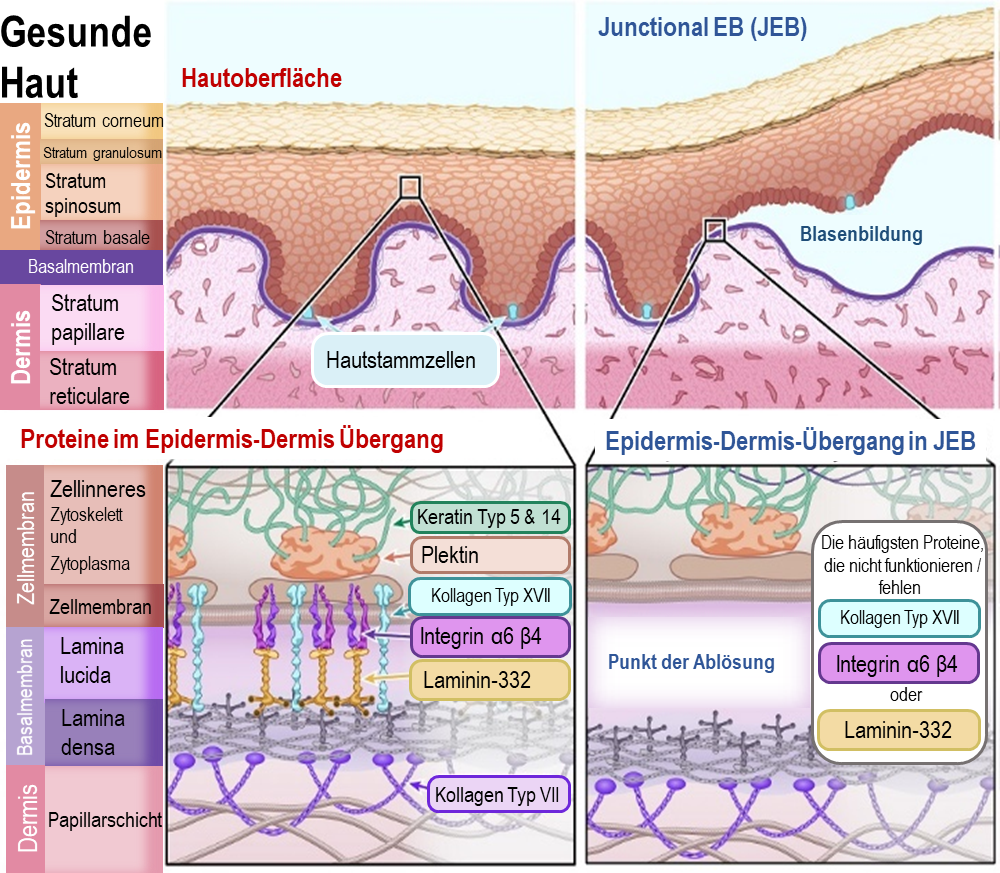
Junktionale EB (JEB) macht etwa 10 % aller EB-Fälle aus. JEB wird als rezessives Merkmal von den Eltern vererbt, was bedeutet, dass eine Person eine EB-assoziierte Genmutation von beiden Elternteilen geerbt haben muss. Diese Eltern hatten wahrscheinlich keine Symptome von EB.
Einige Menschen, die von JEB betroffen sind, leiden unter „schwerer JEB” (früher als generalisierte schwere JEB oder Herlitz-JEB bezeichnet). Die Blasenbildung betrifft den gesamten Körper, einschließlich Mund, Nase und Rachen. Diese Blasenbildung hindert Neugeborene daran, richtig zu essen und zu atmen, was zu Unterernährung und Lungenversagen führt. Tragischerweise ist diese extreme Unterform der JEB lebensbedrohlich, und nur sehr wenige Kinder mit schwerer JEB werden älter als zwei Jahre.
Die andere wesentliche Unterform ist die „intermediäre JEB” (früher als generalisierte intermediäre JEB oder Nicht-Herlitz-JEB bezeichnet). Die Symptome dieser Gruppe sind weniger schwerwiegend, aber dennoch intensiv, und leider sind Todesfälle immer noch häufig. Personen mit intermediärer JEB können bis ins Erwachsenenalter leben, haben jedoch oft starke Vernarbungen, Probleme mit Nägeln und Zähnen sowie andere lebensverkürzende Komplikationen.
Bei JEB lösen sich Epidermis und Dermis an der Basalmembran, genauer gesagt an der Teilschicht Lamina lucida . Diese Teilschicht besteht aus Proteinen, welche die Basalzellen der Epidermis mit der Dermis verankern. Wenn Proteine in dieser Schicht fehlen oder nicht funktionsfähig sind, hält diese Verbindungsstelle der Basalmembran nicht zusammen.
Die wichtigsten Proteine, die bei JEB von Genmutationen betroffen sind, sind:
Kollagen XVII (auch BPAG2 genannt) - [Name des Gens: COL17A1]
Integrin α6β4 (Untereinheiten alpha 6, beta 4) - [Name des Gens: ITGA6 und ITGB4]
Laminin-332 (auch Laminin-5 genannt) - Name des Gens: LAMA3, LAMB3 und LAMC2]
Illustration 3: Diagramm der gesunde Haut und mit Epidermolysis Bullosajunctionalis (EBJ)
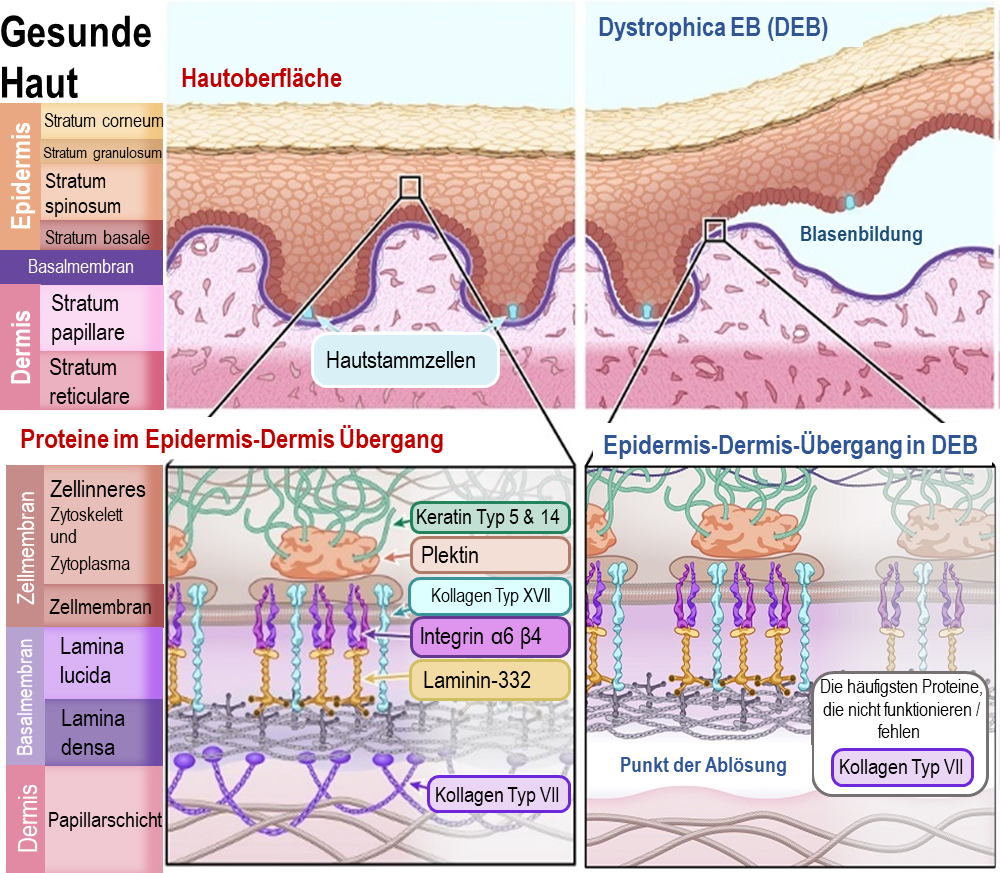
Dystrophe EB
Dystrophe EB (DEB) macht etwa 20 % aller EB-Fälle aus. DEB kann je nach spezifischer Genmutation sowohl dominant als auch rezessiv vererbt werden.
Die Schwere der Symptome variiert bei DEB stark. In vielen Fällen verursacht die dominante Form der DEB leichte Blasenbildung und Symptome, sodass die Betroffenen ein Leben mit leichten Beeinträchtigungen führen können. Die meisten Menschen mit der rezessiven Form der DEB müssen mit weitaus mehr Einschränkungen leben, da die Blasenbildung und Narbenbildung viel stärker ausgeprägt sind und ein erhöhtes Hautkrebsrisiko besteht. Weitere Symptome können Entstellungen sein, die bei der Heilung von Blasen und Wunden entstehen. Diese beinhalten zum Beispiel das Zusammenwachsen von Fingern oder Zehen, Bewegungseinschränkungen der Gelenke und Verengungen der Speiseröhre, die das Schlucken erschweren.
Bei DEB lösen sich Epidermis und Dermis unterhalb der Basalmembran voneinander. Das Protein, welches die Basalmembran hauptsächlich mit der Dermis verbindet, ist Kollagen VII. Dieses Protein bildet Ankerfibrillen, welche in die strukturellen Proteine der Dermis vernetzt sind. Ohne Kollagen VII löst sich die Basalmembran einfach von der Dermis ab.
Die wichtigsten Proteine, die bei DEB von Genmutationen betroffen sind, sind:
Kollagen VII - [Name des Gens: COL7A1]
Illustration 4: Diagramm der gesunde Haut und mit Dystrophica Epidermolysis Bullosa (EBD)
Kindler-Syndrom
Kindler-syndrom (KEB) ist eine seltene Unterform der erblichen EB. KEB ist eine rezessive Form der EB, was bedeutet, dass eine Person die Genmutation von beiden Elternteilen erbt. Die Symptome können von leicht bis schwer reichen und umfassen empfindliche Haut, Lichtempfindlichkeit, Verfärbungen und Verdickungen der Haut. Zahnfleischerkrankungen, Blasen im Mund und ein entzündeter Darm können das Essen beeinträchtigen, und Menschen mit KEB haben ein erhöhtes Risiko, an Hautkrebs zu erkranken.
KEB kann verschiedene und mehrere Hautschichten betreffen. Fehlen die betroffenen Proteine oder sind nicht funktionsfähig, dann wird das Wachstum und die Teilung sowie die Fähigkeit der Keratinozyten (Hautzellen) Epidermis und Dermis miteinander zu verbinden gestört.
Die wichtigsten Proteine, die bei KEB von Genmutationen betroffen sind, sind:
Kindlin-1 - [Name des Gens:FERMT]
Unsere Partner im EB Research Network haben weitere Informationen über EB und ihre Formen
Derzeit gibt es keine zugelassenen Therapien für EB, welche die zugrundeliegenden EB-gekoppelten genetischen Mutationen korrigieren. Behandlungen fokussieren sich vor Allem darauf, die Symptome von EB zu verhindern oder zu lindern. Mögliche Präventivmaßnahmen sind das Umwickeln oder Polstern der Haut um Reibung, Beulen und Verletzungen zu minimieren. Behandlungen können auch auf die Verhinderung und Behandlung von Infektionen und Heilung von Blasen und Wunden abzielen.
Bei schweren Fällen von EB können Medikamente eingesetzt werden, um Infektionen offener Wunden zu verhindern. Operationen können durchgeführt werden um zu verhindern, dass Finger oder Zehen zusammenwachsen oder sich Rachen und Speiseröhre verengen.
In extremen Fällen von EB decken Ärzte große Wunden mit Hauttransplantationen ab. Das Einsetzen von Transplantaten ist jedoch ein seltenes Verfahren, da das Immunsystem in den meisten Fällen das gespendete Transplantat abstoßen wird, selbst wenn Patient und Spender stark miteinander übereinstimmen. Deshalb nutzen Ärzte häufig temporären Hautersatz um große Wunden abzudecken. Auch neue „biologische Hautersatzstoffe“ werden immer häufiger eingesetzt. Diese fortschrittlichen Hautersatzstoffe sollen die Wundheilung verbessern, indem sie synthetische Netze, auf denen Zellen gut wachsen, Proteine aus der Haut und manchmal sogar lebende Keratinozyten verwenden.
Gentherapie bei dystrophe EB
Derzeit ist in der EU eine Gentherapie zugelassen, die auf die genetische Ursache der dystrophischen EB abzielt. Vyjuvek ist eine topische Gentherapie, d. h. sie wird in Form eines Gels direkt auf die Haut aufgetragen. Sie wird zur Heilung der durch EB verursachten Wunden und Läsionen eingesetzt.
Vyjuvek wirkt, indem es zwei gesunde Kopien des COL7A1-Gens an Keratinozyten und Fibroblasten in der Haut abgibt, wodurch diese Kollagen produzieren können und die Wundheilung unterstützt wird. Vyjuvek bringt das COL7A1-Gen mithilfe einer inaktivierten Form des Herpes-simplex-Virus (HSV-1) als Vektor in die Zellen ein. HSV-1 ist dasselbe Virus, das Lippenherpes verursacht. Diese Viruspartikel wurden jedoch so verändert, dass sie sich nicht vermehren können, wodurch sie sicher in der Anwendung sind und keine Infektionen verursachen.
HSV-1 kann die Hautbarriere nicht passieren, sodass diese Behandlung nur bei offenen Wunden oder Läsionen angewendet werden kann. Das bedeutet, dass sie nur die Wundheilung unterstützen kann, aber nicht deren Entstehung verhindern kann. Die Behandlung ist nicht dauerhaft, sodass eine kontinuierliche Anwendung über einen längeren Zeitraum erforderlich ist, um die klinische Wirksamkeit aufrechtzuerhalten.
Derzeit ist in der EU eine Gentherapie zugelassen, die auf die genetische Ursache der dystrophischen EB abzielt. Vyjuvek ist eine topische Gentherapie, d. h. sie wird in Form eines Gels direkt auf die Haut aufgetragen. Sie wird zur Heilung der durch EB verursachten Wunden und Läsionen eingesetzt.
Vyjuvek wirkt, indem es zwei gesunde Kopien des COL7A1-Gens an Keratinozyten und Fibroblasten in der Haut abgibt, wodurch diese Kollagen produzieren können und die Wundheilung unterstützt wird. Vyjuvek bringt das COL7A1-Gen mithilfe einer inaktivierten Form des Herpes-simplex-Virus (HSV-1) als Vektor in die Zellen ein. HSV-1 ist dasselbe Virus, das Lippenherpes verursacht. Diese Viruspartikel wurden jedoch so verändert, dass sie sich nicht vermehren können, wodurch sie sicher in der Anwendung sind und keine Infektionen verursachen.
HSV-1 kann die Hautbarriere nicht passieren, sodass diese Behandlung nur bei offenen Wunden oder Läsionen angewendet werden kann. Das bedeutet, dass sie nur die Wundheilung unterstützen kann, aber nicht deren Entstehung verhindern kann. Die Behandlung ist nicht dauerhaft, sodass eine kontinuierliche Anwendung über einen längeren Zeitraum erforderlich ist, um die klinische Wirksamkeit aufrechtzuerhalten.
Aktuelle Forschung
Verbesserung der Lebensqualität
Zahlreiche Forscher, Pharmaunternehmen und Hersteller medizinischer Produkte arbeiten daran, bessere Produkte zur Linderung der Symptome von EB zu entwickeln.
Die untersuchten Produkte reichen von besseren Verbänden und Bandagen bis hin zu Salben und Lotionen, die die Hautheilung unterstützen.
“Neue” Haut herstellen
Der ideale Weg eine genetische Erkrankung zu heilen ist die Korrektur oder das Ersetzen des mutierten Gens in der DNA einer Person. Wissenschaftler entwickeln “Gentherapien” die genau dies tun.
Neue Entdeckungen und Technologien, wie zum Beispiel CRISPR-Cas9 Geneditierung, beschleunigen den Prozess bei dem DNA und Gene innerhalb der Zelle verändert werden können. Jedoch ist die Veränderung der DNA einer Person keine einfache Aufgabe. Der Prozess kann außerdem Risiken mit sich bringen. Wenn beim Hinzufügen oder Verändern eines Gens ein Fehler passiert, können Krebs, neue Komplikationen oder eine Verschlechterung des Zustands der Person die Folge sein. Klinische Studien sind wichtig, um Risiken neuer Behandlungen zu identifizieren und zu zeigen, dass sie tatsächlich funktionieren.
In vielen Fällen geht der Einsatz von Stammzellen mit Gentherapien einher. Für EB Gentherapien wollen Wissenschaftler die DNA von Hautstammzellen anstatt anderer Hautzelltypen verändern, denn Hinzufügen (oder Veränderung) eines Gens in einer Hautstammzelle verursacht das Weitergeben dieses Gens an alle anderen Zellen der Epidermis, die die Stammzellen herstellen. Das ist wichtig, da alle Keratinozyten unserer Haut alle 4-5 Wochen ersetzt werden. Außerdem sind Hautstammzellen in der Lage sich selbst zu erneuern, sodass alle Veränderungen der DNA potenziell “permanent” sein könnten (oder zumindest solange bestehen, wie die Stammzelle überlebt).
In mehreren klinischen Studien wird die Veränderung der DNA von Patientenhautstammzellen um „neue“ Haut für Transplantationen herzustellen getestet. Obwohl die Methoden und die Details verschiedener Studien variieren, sind die Idee und der generelle Prozess gleich. Er benötigt das Abnehmen von Hautproben einer Person, die an EB erkrankt ist, und die Kultivierung dieser Zellen im Labor in der Hoffnung, dass diese Proben Hautstammzellen beinhalten. Methoden der DNA- Veränderung werden dann genutzt um Gene innerhalb der Zelle hinzuzufügen oder zu verändern um so eine normale Proteinproduktion wiederherzustellen. Die Hautstammzellen mit den neuen DNA-Änderungen werden dann mittels fortgeschrittener Zellkulturmethoden genutzt, um eine neue Hautschicht im Labor heranzuziehen. Diese Labor-gezüchtete Hautschicht kann dann in den Patienten eingepflanzt werden.
Manche dieser Methoden nutzen induzierte pluripotente Stammzellen (iPSC), ein Stammzelltyp der von Zellen aus unserem Körper hergestellt werden kann. Diese Zellen können sich unendlich lange teilen und jeden anderen Zelltyp unserer Haut herstellen. Sie könnten also eine Quelle für Zellen, die für die Ersetzung in zerstörtem Gewebe gebraucht werden, darstellen.
Diese Art der Behandlung benötigt viel Zeit und Arbeit um die genetische Mutation eines EB Patienten zu identifizieren, ein spezifisches DNA-Veränderungsverfahren zu entwickeln, mehrere Wochen die es benötigt die Zellen wachsen zu lassen und eine OP, bei der die Hauttransplantation durchgeführt wird. Außerdem werden spezialisiert Forschungs- und Zellkultureinrichtungen benötigt. Trotz dieser Hindernisse wurden zuletzt große Fortschritte erzielt, die zeigen, dass der Prozess funktionieren kann. Der folgende Abschnitt beschreibt eine dieser Erfolgsgeschichten.
Rampenlicht auf europäische Forschung: Eine Erfolgsgeschichte
Im Jahr 2017 berichteteeine Gruppe von Wissenschaftlern und Ärzten, geleitet von Professor Michele De Luca, den erfolgreichen Einsatz einer Gentherapie und Labor-gezüchteter Hauttransplantate um einen Jungen zu retten, der über 80% seiner Haut verloren hatte. Der Junge war an JEB erkrankt, ausgelöst durch Mutationen im LAMB3 Gen, die er von beiden Elternteilen geerbt hat. Alle Behandlungen, inklusive mehrerer extremer Verfahren, sind gescheitert und seine Ärzte waren sich einig, dass der Junge wahrscheinlich nicht überleben wird. Regierungsvertreter haben die Arzneimittel-Härtefall („compassionate use“)für eine neuartige Gentherapie erteilt,, welche zuvor erst in zwei Fallstudien getestet wurde, welche jeweils nur einen Patienten untersucht haben. Die Eltern des Jungen haben der Prozedur zugestimmt, trotz Warnungen, dass ihr Sohn die Prozedur womöglich nicht überleben wird.
Wissenschaftler begannen Hautproben des Jungen zu nehmen und die Zellen im Labor zu kultivieren. Sie hofften darauf Zellanhäufungen zu entnehmen, die Hautstammzellen beinhalten. Diese Zellhaufen wurden dann mit einem Virus behandelt, welches eine funktionstüchtige Kopie des LAMP3 Gens in die DNA der Hautstammzellen einfügte. Dieser Prozess repariert nicht die Mutation, die der Junge in seinen eigenen Kopien des Gens besaß. Stattdessen fügt er neue DNA mit einer Kopie des LAMB3 Gens in die Hautstammzellen des Jungen. Das neue Gen erlaubt es den Zellen Laminin Proteine herzustellen, die funktionsfähig sind. Die Hautstammzellen mit dem funktionierenden LAMB3 Gen wurden dann genutzt um Hautschichten im Labor zu züchten, wobei fortgeschrittene Zellkulturtechniken angewandt wurden. Dieser Prozess dauerte mehrere Wochen.
Die kleinen Stücke Labor-gezüchteter Haut wurden dann in zwei separaten Prozeduren im Abstand von einem Monat auf den Jungen transplantiert.
Am Ende hat der Junge überlebt und lebt nun mit einer Haut die zu 80% Gen-veränderten Zellen besteht. Die Wissenschaftler haben die Haut untersucht und berichten, dass die Haut auf viele Weisen erstaunlich wie normale Haut aussieht.
Der Erfolg dieser Behandlung ist sehr spannend; manche sagen sogar die Ergebnisse seien ein Wunder, weil sie die Erwartungen übertroffen haben.
Leider sind Gentherapien wie diese noch nicht klinisch zugelassen oder stehen zur breiteren Verfügung. Personalisierte Medizin wie diese sind kostenaufwändig, benötigen spezielle Einrichtungen und den Einsatz vieler Menschen. Hoffentlich werden Behandlungen wie diese in der Zukunft zugelassen, bezahlbar und breiter verfügbar für die vielen Formen von EB.
Die Zukunft ist vielversprechend. Die oben beschriebene Erfolgsgeschichte sowie positive Ergebnisse anderer Studien haben zur Durchführung weiterer klinischer Studien angeregt. M. De Luca vom Center für regenerative Medizin “Stefano Ferrari” in Moderna, Italien, gemeinsam mit JW Bauer vom Universitätsklinikum für Dermatologie in Salzburg, Österreich leiten mehrere klinische Studien, welche ähnliche aber weiter entwickelt Prozeduren verwenden um Mutationen in den COL7A1 und COL17A1Genen zu ersetzen (Studie1, Studie2). Wissenschaftler an anderen Instituten entwickeln ebenfalls ihre eigenen Gentherapien, wie zum Beispiel die klinische Studie unter der Leitung von Jean Yuh Tang von der Stanford Universität für Medizin, welche ebenfalls auf die zugrundeliegenden Mutationen im COL7A1 Gen, welche zu DEB führt, abzielt.
Weitere Behandlungsmethoden
Wie im vorherigen Abschnitt erwähnt sind Methoden zur Herstellung DNA-veränderter Haut sehr herausfordernd; der Prozess ist komplex, kostenaufwändig und hat Risiken. Außerdem kann es mehrere Jahre dauern bis diese Methode zu einer realistischen Behandlungsoption wird.
Diese Herausforderungen haben Wissenschaftler dazu veranlasst über weitere Behandlungsmethoden für EB nachzudenken, die möglicherweise schneller realisierbar, kostengünstiger und schneller klinisch zugelassen werden könnten. Beispiele sind Stammzellen, Medikamente, Pflanzenextrakte und andere Behandlungen. Die beiden unten beschrieben sind nur zwei Beispiele dieser Methoden.
Therapeutische Cremes und Lotionen zur äußerlichen Anwendung werden entwickelt, um den Problemen, die durch EB Mutationen entstehen, entgegenzuwirken. Eine topische Behandlung, Vyjuvek, wurde bereits für die Verwendung in der EU zugelassen. Eine besonders interessante klinische Studie untersucht eine Creme, die versucht, das COL7A1-Gen zu reparieren, was Menschen mit dystropher EB helfen würde. Die Creme enthält Inhaltsstoffe, die die Hautzellen dazu anregen, die Mutation im COL7A1-Gen bei der Bildung des Kollagen-VII-Proteins zu überspringen.
Knochenmarktransplantationen werden zur Behandlung von Leukämie und anderen Erkrankungen des Blut- und Immunsystems eingesetzt. Jedoch untersuchen einige Forschungsgruppen die Behandlung von EB mit allogenen Knochenmarkstransplantationen, bei denen Stammzellen aus einem gesunden Spender entnommen werden. Der Vorteil dieser Methode ist, dass Knochenmarkstransplante für viele Jahre erfolgreich genutzt wurden und die zugrundeliegende Technik während dieser Zeit sehr verbessert wurde. Erste Ergebnisse zeigen, dass gesunde Stammzellen aus dem Knochenmark (oder Zellen die aus diesen Stammzellen enstehen) Proteine des Bindegewebes herstellen, wie zum Beispiel Kollagen VII, welche bei DEB Patienten fehlen. Trotzdem hat diese Methode ihre Einschränkungen, vor allem die Abhängigkeit vom Auffinden eines passenden Spenders und die zur Zeit hohen Risiken die während der Prozedur auftreten können.
Eine umfassende Liste klinischer Studien für EB kann unter clinicaltrials.gov eingesehen werden (Bitte beachten Sie, dass diese öffentliche Website nur klinische Studien auflistet. Sie überprüft nicht, ob die aufgeführten klinischen Studien sicher und wissenschaftlich fundiert sind oder von seriösen Einrichtungen durchgeführt werden. Bitte lesen Sie alle Haftungsausschlüsse, sprechen Sie mit vertrauenswürdigen Gesundheitsdienstleistern und informieren Sie sich über mögliche Risiken und Nutzen.
Erfahren Sie mehr
Informationsmaterialien von DEBRA, einer Interessensgruppe für EB Patienten
EB Forschungsnetzwerk: Ein Zusammenschluss von gemeinnützigen Vereinen für die Unterstützung von EB Forschung, angeführt von DEBRA Österreich
Epidermolysis bullosa wird manchmal auch „Schmetterlingshaut” genannt, da die Haut empfindlich ist und leicht verletzt werden kann, ähnlich wie die Flügel eines Schmetterlings.
EB wurde von Heinrich Koebner, einem 1838 geborenen deutschen Dermatologen, benannt.