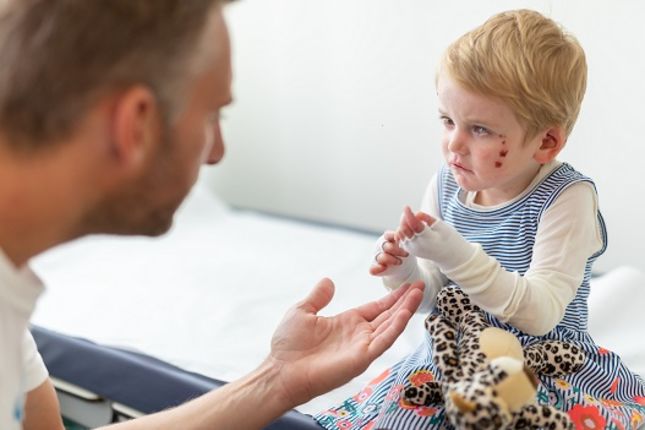
L'epidermolisi bollosa (EB) è una malattia genetica che altera la coesione delle cellule e dei tessuti. Esistono diversi tipi di EB, ma un sintomo comune a tutte le forme è la pelle estremamente fragile che si lacera facilmente o sviluppa piaghe e vesciche anche in seguito a lievi frizioni o lesioni minori. Attualmente non esiste una cura per l'EB, ma la terapia genica è già utilizzata per trattare persone affette da un tipo specifico di EB, mentre ricerche e studi clinici promettenti stanno esplorando nuovi modi in cui la terapia genica e cellulare può essere d'aiuto.
Questa risorsa è stata tradotta automaticamente dall'inglese ed è in attesa di revisione.
Cosa sappiamo?
Tutte le forme di EB sono causate da mutazioni genetiche che influenzano il modo in cui le cellule e i tessuti si connettono tra loro. L'EB semplice, l'EB giunzionale, l'EB distrofica e l'EB di Kindler sono i quattro tipi principali di EB.
L'EB è relativamente rara, con un'incidenza stimata di 1 caso ogni 50.000 nati vivi.
L'EB colpisce visibilmente la pelle, ma questa malattia può anche causare complicazioni in altri organi, come vesciche e piaghe nella gola, nelle vie respiratorie superiori e nelle vie urinarie. Può provocare una malattia sistemica progressiva con conseguenze che limitano la vita e può portare a tumori cutanei aggressivi.
Attualmente non esiste una cura per l'EB, ma recentemente è stata approvata una terapia genica topica per aiutare a trattare le ferite causate da un tipo specifico di EB.
A cosa stanno lavorando i ricercatori?
Sono molti i nuovi dispositivi, farmaci e trattamenti biologici attualmente in fase di sperimentazione clinica. Molti di questi studi mirano a migliorare la gestione dell'EB. Alcuni studi mirano a “correggere” le mutazioni genetiche alla base dell'EB.
Lo sviluppo di nuove terapie geniche potrebbe portare alla possibilità di ripristinare i geni sani nelle cellule della pelle. Anche il lavoro con le cellule staminali epidermiche (della pelle), l'ottimizzazione delle tecniche di coltivazione della pelle in laboratorio e il miglioramento dei metodi di innesto dei tessuti sono componenti fondamentali per lo sviluppo di terapie geniche efficaci per l'EB
Quali sono le sfide?
Il trattamento di una malattia genetica è molto difficile perché la causa risiede nel DNA di una persona. I trattamenti per compensare le mutazioni genetiche devono riparare il gene mutato o aggiungere una nuova copia funzionale del gene alle cellule. Non è un compito semplice. Modificare il DNA cellulare o aggiungere nuovi geni alle cellule comporta potenziali rischi, come la possibilità di sviluppare un tumore. I trapianti di cellule e di pelle possono comportare rischi quali emorragie, infezioni e rigetto dell'innesto.
Un'attenta ricerca clinica mira a garantire che i nuovi trattamenti per l'EB siano sicuri ed efficaci.
Una sfida ancora da affrontare è quella di rendere i trattamenti complessi, come le terapie geniche, accessibili e alla portata di tutti.
Informazioni sull'epidermolisi bollosa
L'epidermolisi bollosa (EB) è un gruppo di rare malattie genetiche del tessuto connettivo che causano una fragilità estrema della pelle con formazione di vesciche. Le vesciche possono essere causate da attrito sulla pelle, piccole lesioni cutanee e attività quotidiane, come sfregamento e graffi.
Queste vesciche possono trasformarsi in piaghe dolorose e ferite aperte. Nelle forme gravi di EB, le vesciche e le piaghe si sviluppano anche in altri tessuti, come la bocca, la gola, lo stomaco, le vie respiratorie superiori e le vie urinarie.
Esiste un'ampia gamma di gravità dell'EB. I tipi lievi di EB possono comportare la comparsa di vesciche minori per tutta la vita, mentre altri presentano sintomi gravi solo durante l'infanzia, che possono migliorare con l'età. Alcune forme di EB possono essere pericolose per la vita a causa della perdita di grandi quantità di pelle. Altre possono causare cicatrici estese, fusione delle dita delle mani e dei piedi e altre complicazioni che limitano la vita. Alcune forme della malattia possono portare allo sviluppo di tumori della pelle, riducendo l'aspettativa di vita.
L'EB è rara, il che rende difficile stimare il numero totale di persone affette da EB. DEBRA, un'organizzazione benefica dedicata all'EB, stima che circa 500.000 persone in tutto il mondo siano affette da una qualche forma di EB.
Tutti i tipi ereditari di EB sono causati da mutazioni nel DNA di una persona. Queste mutazioni alterano uno dei diversi geni importanti per le proteine che collegano le cellule e i tessuti tra loro. Ecco perché l'EB è anche definita una malattia del “tessuto connettivo”. Attualmente nel nostro DNA sono stati individuati 16 geni collegati all'EB ereditaria classica.
Come si trasmette l'EB? Una mutazione genica legata all'EB viene spesso trasmessa dai genitori ai figli. Il nostro DNA contiene due copie di ogni gene (ad eccezione dei geni localizzati sui cromosomi X e Y). Diverse forme di EB richiedono l'eredità di una sola copia di una mutazione genica legata all'EB. Queste sono definite forme di EB a carattere "dominante". Il genitore che trasmette una mutazione genetica dominante legata all'EB ha anch'esso l'EB e i suoi sintomi. Altre forme della patologia richiedono l'eredità di una mutazione genetica associata all’ EB da entrambi i genitori. Queste forme sono chiamate "recessive". Le forme recessive di EB sono spesso una sorpresa per una famiglia, perché nessuno dei due genitori presenta i sintomi della patologia e potrebbero non sapere di essere portatori della mutazione.
La pelle
La pelle è un organo esteso e complesso. Funge da scudo resistente, flessibile e impermeabile che trattiene l'acqua e tiene lontani i batteri. Ci protegge dal vento e dalle intemperie e ci aiuta a regolare la temperatura corporea. È il luogo in cui viene sintetizzata la vitamina D. Oltre a tutto questo, la nostra pelle ha un numero enorme di cellule specializzate che ci permettono di percepire la temperatura, le consistenze, la pressione e il dolore.
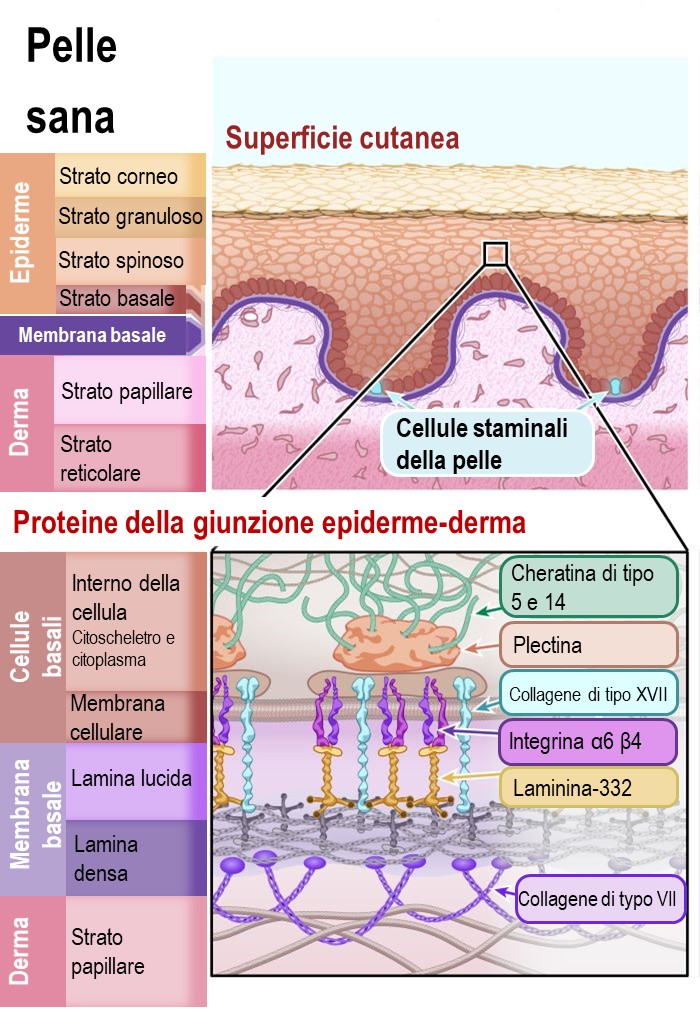
I due strati principali della pelle sono l'epidermide (lo strato più esterno) e il derma. I ricercatori suddividono ulteriormente questi strati. È possibile vedere questi sottostrati nel diagramma sottostante.
L'epidermide è costituita principalmente da cellule chiamate cheratinociti. Sono chiamati cheratinociti per via della proteina resistente che producono, chiamata cheratina, la stessa proteina che costituisce i nostri capelli e le nostre unghie. I cheratinociti sono prodotti da cellule staminali della pelleche si trovano nel sottostrato dello strato basale dell'epidermide e nei follicoli piliferi del derma. Le cellule staminali della pelle producono costantemente nuovi cheratinociti per sostituire le cellule cutanee che cadono o si sfregiano continuamente. Questo rende le cellule staminali della pelle molto importanti per mantenere la salute della nostra pelle.
Il derma è lo strato della pelle che contiene i vasi sanguigni, le cellule sensoriali per il tatto, la temperatura e il dolore, le ghiandole sudoripare, i follicoli piliferi e altro ancora. Nel derma sono presenti molti tipi diversi di cellule, ma la maggior parte del derma è in realtà costituito da una rete flessibile di fibre connettive e proteine. Questa rete di proteine sostiene la struttura della pelle e mantiene tutto al suo posto. Le proteine coinvolte in questa rete sono principalmente il collagene e l'elastina.
Anche il punto di connessione tra l'epidermide e il derma è importante, in particolare quando si parla di EB. La “membrana basale” è un sottile strato di tessuto connettivo importante per legare insieme l'epidermide e il derma. La membrana basale è costituita principalmente da due tipi di proteine, la laminina e il collagene.
Esistono quattro tipi principali di EB: EB semplice (EBS), EB giunzionale (JEB), EB distrofica (DEB) e EB di Kindler (KEB). Queste categorie sono comunemente distinte in base allo strato di pelle in cui si formano le vesciche. Sebbene tutte le forme di EB abbiano come sintomo la formazione di vesciche, queste non sono causate dallo stesso problema. Il gene colpito da una mutazione genetica legata all'EB determina dove, perché e in che misura si verificano le vesciche e il distacco della pelle. Determina anche il tipo di EB di cui una persona è affetta.
Attualmente nel nostro DNA sono stati individuati 16 geni specifici collegati all'EB classica. Le mutazioni di questi geni impediscono alle cellule di produrre importanti proteine funzionali che tengono insieme l'epidermide e il derma. Conoscere il ruolo che le proteine svolgono normalmente nel tenere insieme la pelle è fondamentale per comprendere i diversi tipi di EB. Ciò rivela anche perché alcuni tipi di EB sono più gravi di altri e perché un trattamento per un tipo di EB potrebbe non funzionare per tutti.
Di seguito sono riportate delle sintesi che evidenziano i geni, le loro proteine e alcune specificità relative a dove e perché si verifica il distacco per tre delle principali forme di EB.
EB Simplex
L'EB semplice (EBS) è la forma più comune di EB e costituisce circa il 70% di tutti i casi di EB. La gravità dei sintomi dell'EBS può variare, da fragilità cutanea e piccole vesciche su mani e piedi, a casi in cui le vesciche si formano su tutto il corpo, fino a sottotipi più gravi. L'EBS è prevalentemente ereditaria dai genitori come tratto dominante, il che significa che è sufficiente che un solo genitore trasmetta la mutazione genetica associata all'EB. Quel genitore ha l'EBS e probabilmente ha (o ha avuto) i sintomi associati.
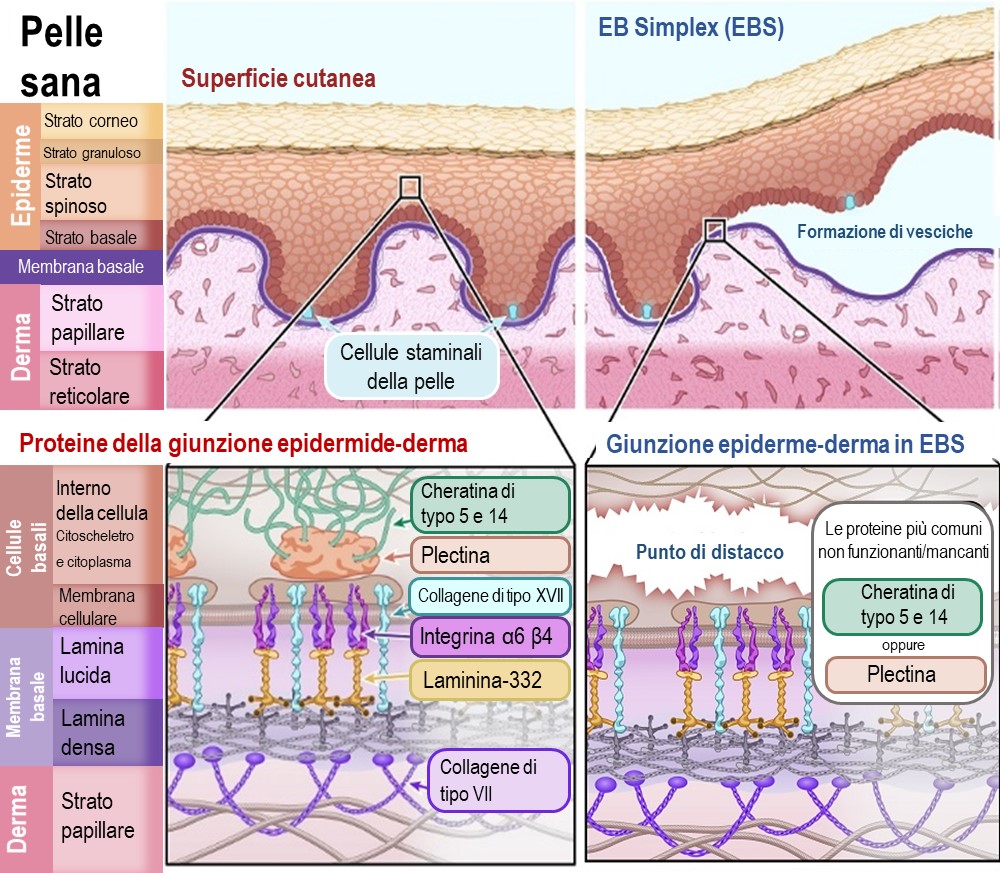
Nell'EBS, la separazione dell'epidermide e del derma avviene all'interno delle cellule basali dell'epidermide (Stratum basale), appena sopra la membrana basale. Il problema è che le proteine che fissano le cellule basali alla membrana basale non si attaccano correttamente al supporto strutturale all'interno della cellula (il citoscheletro). Questo provoca la lacerazione e la rottura delle cellule basali, lasciando la parte inferiore di queste cellule attaccata alla membrana basale.
Le proteine primarie colpite dalle mutazioni genetiche nell'EBS sono:
Cheratina tipo 5 – [Nome del gene: KRT5]
Cheratina tipo 14 – [Nome del gene: KRT14]
Plectina – [Nome del gene: PLEC1]
Illustrazione 2 : Schema di pelle sana e pelle affetta da epidermolisi bollosa simplex (EBS)
EB giunzionale
L'EB giunzionale (EBG) costituisce circa il 10% di tutti i casi di EB. La EBG è ereditaria dai genitori come tratto recessivo, il che significa che un individuo deve aver ereditato una mutazione genetica legata all'EB da entrambi i genitori. È probabile che i genitori non presentassero alcun sintomo di EB.
Alcune persone affette da EBG presentano una forma “grave” (precedentemente denominata EBG generalizzata grave o EBG di Herlitz). Le vesciche sono presenti su tutto il corpo, compresi bocca, naso e gola. Queste vesciche impediscono ai neonati di nutrirsi e respirare correttamente, causando malnutrizione e insufficienza polmonare. Purtroppo, questo sottotipo estremo di EBG è potenzialmente letale e pochissimi bambini affetti da EBG grave vivono oltre i due anni di età.
L'altro sottotipo principale è la “EBG intermedia” (precedentemente chiamata EBG generalizzata intermedia o EBG non Herlitz). I sintomi di questo gruppo sono meno gravi, ma comunque intensi e, purtroppo, i decessi sono ancora frequenti. Gli individui affetti da EBG intermedia possono vivere fino all'età adulta, ma spesso presentano cicatrici estese, problemi alle unghie e ai denti, nonché altre complicanze che limitano la loro aspettativa di vita.
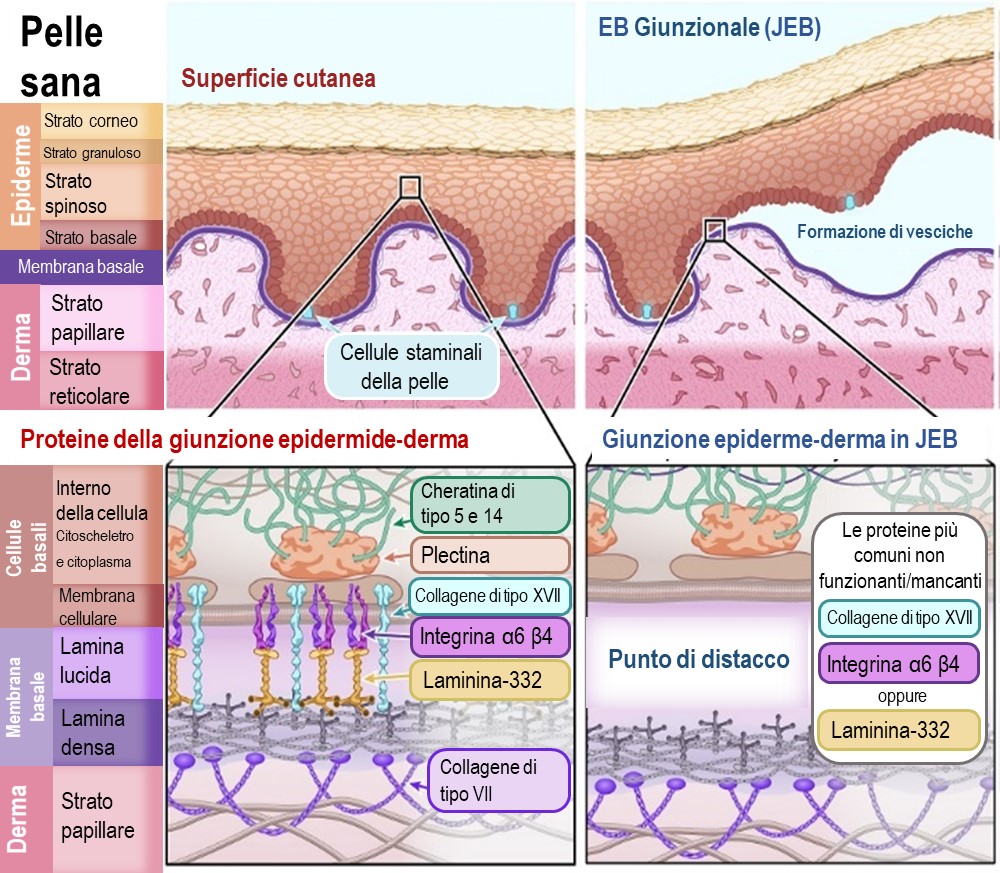
Nella EBG, la separazione dell'epidermide e del derma avviene a livello della membrana basale, in particolare nel sottostrato della “lamina lucida”. Questo sottostrato è costituito da proteine che ancorano le cellule basali dell'epidermide al derma. Quando le proteine di questo strato sono assenti o non funzionano, questo punto di giunzione della membrana basale non riesce a tenere insieme le cellule.
Le proteine primarie colpite dalle mutazioni genetiche nella EBG sono:
Collagene di tipo XVII (noto anche come BPAG2) – [Nome del gene:COL17A1]
Integrina α6β4 (subunità alfa 6, beta 4) – [Nomi dei geni: ITGA6 e ITGB4]
Laminina-332 (nota anche come Laminina-5) – [Nomi dei geni: LAMA3, LAMB3 e LAMC2]
Illustrazione 3 : Schema di pelle sana e pelle affetta da epidermolisi bollosa giunzionale (JEB)
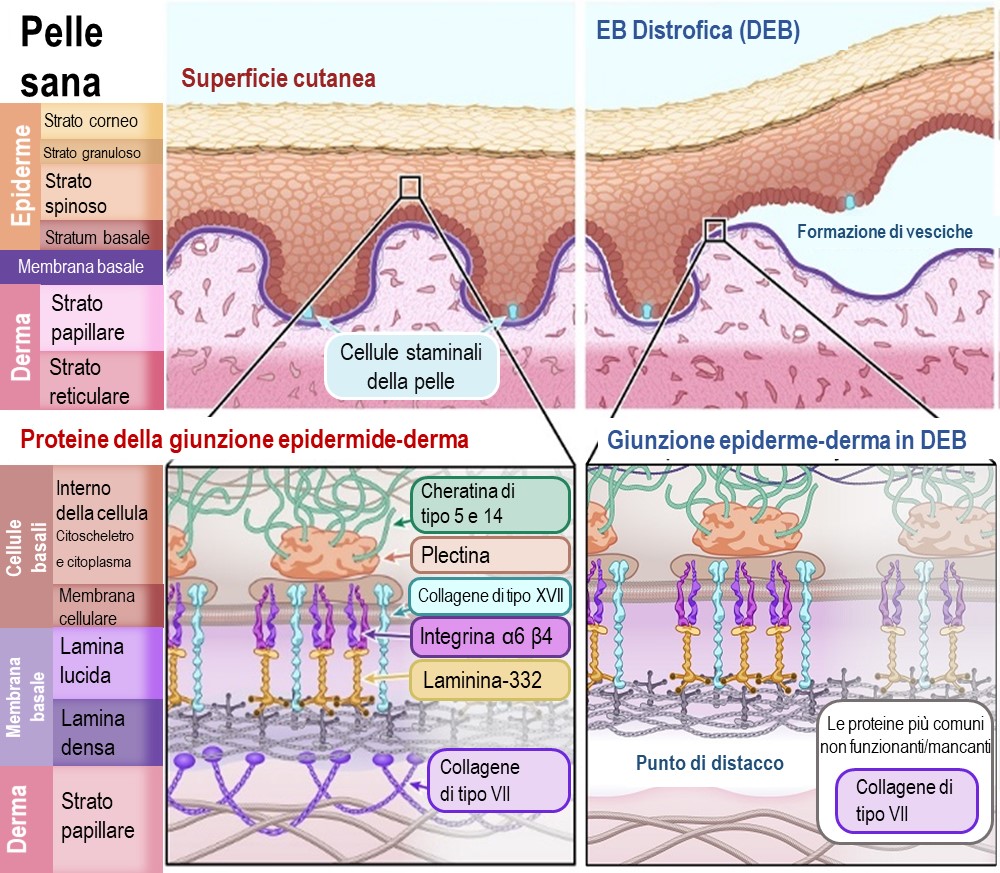
EB distrofica
L'EB distrofica (EBD) costituisce circa il 20% di tutti i casi di EB. La EBD può essere ereditaria sia come tratto dominante che recessivo, a seconda della mutazione genetica specifica.
La gravità dei sintomi varia notevolmente per la EBD. In molti casi, la forma dominante della EBD causa lievi vesciche e sintomi, che consentono a questi individui di condurre una vita con lievi disabilità. La maggior parte delle persone affette dalla forma recessiva della EBD deve affrontare molte più limitazioni, poiché le vesciche e le cicatrici sono molto più gravi e vi è un aumento del rischio di cancro della pelle. Altri sintomi possono includere deturpazioni dovute alla guarigione delle vesciche e delle ferite, fusione delle dita delle mani e dei piedi, limitazione dei movimenti articolari e restringimento dell'esofago (tubo che trasporta il cibo), con conseguente difficoltà a deglutire.
Nella DEB, la separazione dell'epidermide e del derma avviene sotto la membrana basale. La proteina primaria che lega la membrana basale al derma è il collagene VII. Questa proteina forma fibrille di ancoraggio che si intrecciano con le proteine strutturali del derma. Senza il collagene VII, la membrana basale si stacca facilmente dal derma.
La proteina primaria colpita da una mutazione genetica nella EBD è:
Collagene di tipo VII – [Nome del gene: COL7A1]
Illustrazione 4 : Schema di pelle sana e pelle affetta da epidermolisi bollosa distrofica (DEB)
EB di Kindler (o Sindrome di Kindler)
L'EB di Kindler (KEB) è un sottotipo raro di EB ereditaria. La KEB è una forma recessiva di EB, il che significa che un individuo eredita la mutazione genetica da entrambi i genitori. I sintomi possono variare da lievi a gravi e comprendono fragilità cutanea, sensibilità alla luce, scolorimento e ispessimento della pelle. Malattie gengivali, vesciche nella bocca e infiammazioni intestinali possono influire sull'alimentazione e le persone affette da KEB hanno un rischio maggiore di sviluppare il cancro della pelle.
La KEB può colpire diversi strati della pelle. Quando la proteina interessata è assente o non funziona correttamente, impedisce ai cheratinociti (cellule della pelle) di crescere e dividersi normalmente e di attaccare l'epidermide al derma.
La proteina principalmente interessata dalla mutazione genetica nella KEB è:
Kindlin-1 – [Nome del gene: FERMT1]
I nostri partner dell'EB Research Network dispongono di informazioni sull'EB e sulle sue forme
I trattamenti si concentrano principalmente sulla prevenzione o sull'alleviamento dei sintomi dell'EB. Le misure preventive possono includere l'uso di bendaggi e imbottiture per ridurre al minimo l'attrito, gli urti e i piccoli traumi alla pelle. I trattamenti possono anche mirare a prevenire le infezioni e aiutare la guarigione delle vesciche e delle piaghe.
Nei casi più gravi di EB, possono essere utilizzati farmaci per prevenire le infezioni delle ferite aperte e delle piaghe. È possibile ricorrere anche alla chirurgia per impedire la fusione delle dita delle mani e dei piedi o il restringimento della gola e dell'esofago (tubo che trasporta il cibo).
In casi estremi di EB, i medici possono coprire le ferite aperte di grandi dimensioni con innesti cutanei. Tuttavia, l'innesto di pelle da donatori è molto raro perché il sistema immunitario rigetta quasi sempre gli innesti cutanei, anche se il paziente e il donatore sono compatibili. Pertanto, i medici utilizzano spesso sostituti cutanei temporanei per coprire grandi ferite aperte. Stanno diventando sempre più comuni anche i nuovi “sostituti cutanei biologici”.
Questi sostituti cutanei avanzati mirano a migliorare la guarigione delle ferite utilizzando reti sintetiche su cui le cellule possono crescere, proteine presenti nella pelle e talvolta anche cheratinociti viventi.
Terapia genica per l'EB distrofica
Attualmente esiste una terapia genica approvata nell'UE che agisce sulla causa genetica alla base dell'EB distrofica. Vyjuvek è una terapia genica topica, il che significa che viene applicata direttamente sulla pelle sotto forma di gel. Viene utilizzata per aiutare a guarire le ferite e le lesioni causate dall'EB.
Vyjuvek agisce trasportando due copie sane del gene COL7A1 ai cheratinociti e ai fibroblasti della pelle, consentendo loro di produrre collagene e favorendo la guarigione delle ferite. Vyjuvek trasporta il gene COL7A1 nelle cellule utilizzando una forma inattiva del virus dell'herpes simplex (HSV-1) come vettore. L'HSV-1 è lo stesso virus che causa l'herpes labiale. Tuttavia, queste particelle virali sono state modificate per impedirne la replicazione, rendendole sicure da usare e impedendo loro di causare infezioni.
Ricerca attuale
Miglioramento della qualità di vita
Molti ricercatori, aziende farmaceutiche e aziende di forniture mediche stanno lavorando allo sviluppo di prodotti migliori per migliorare la gestione dei sintomi dell'EB.
I prodotti in fase di studio vanno da cerotti e bendaggi migliori a pomate e lozioni che aiutano la pelle a guarire.
Produrre "nuova" pelle
Il modo ideale per correggere una malattia genetica è correggere o sostituire un gene mutato nel DNA di una persona. I ricercatori stanno creando terapie geniche che fanno proprio questo. Nuove scoperte e tecnologie, come l'editing genetico CRISPR-Cas9, stanno accelerando notevolmente il modo in cui il DNA e i geni possono essere modificati nelle cellule. Tuttavia, modificare il DNA delle cellule di una persona non è un compito semplice. Il processo potrebbe anche comportare gravi rischi. Se l'aggiunta o la modifica di un gene non va a buon fine, può causare il cancro, aggiungere nuove complicazioni o peggiorare le condizioni di una persona. Gli studi clinici sono importanti per identificare i rischi dei nuovi trattamenti e dimostrare che funzionano davvero.
In molti casi, le cellule staminali vanno di pari passo con le terapie geniche. Per le terapie geniche dell'EB, i ricercatori vogliono modificare il DNA delle cellule staminali della pelle, piuttosto che altri tipi di cellule cutanee. Questo perché l'aggiunta (o la modifica) di un gene nelle cellule staminali della pelle farà sì che quel gene venga trasmesso a tutte le altre cellule prodotte dalle cellule staminali. Questo è importante perché quasi tutti i cheratinociti della nostra pelle vengono sostituiti ogni 4-5 settimane. Inoltre, le cellule staminali della pelle si rinnovano continuamente, quindi qualsiasi modifica del DNA introdotta in una cellula staminale potrebbe essere potenzialmente “permanente” (o durare almeno finché le cellule staminali sopravvivono).
Diversi studi clinici stanno testando metodi per modificare il DNA delle cellule staminali della pelle dei pazienti affetti da EB al fine di far crescere “nuova” pelle sana da trapiantare. Sebbene i metodi e i dettagli di questi diversi studi clinici variano, l'idea e il processo generale sono sostanzialmente gli stessi. Vengono prelevati campioni di pelle da una persona affetta da EB e coltivati in laboratorio; si spera che questi campioni contengano cellule staminali della pelle. Vengono quindi utilizzati metodi di modifica del DNA per aggiungere o modificare i geni nelle cellule e ripristinare la normale produzione di proteine. Le cellule staminali della pelle con le nuove modifiche del DNA vengono quindi utilizzate in metodi avanzati di coltura cellulare per far crescere strati di epidermide in laboratorio. Questi strati coltivati in laboratorio possono poi essere innestati sul paziente.
Alcuni di questi approcci prevedono l'uso di cellule staminali pluripotenti indotte (iPSC), un tipo di cellule staminali che possono essere generate dalle cellule del nostro corpo. Queste cellule possono propagarsi (auto-rinnovarsi) indefinitamente e possono dare origine a tutti gli altri tipi di cellule della pelle. Potrebbero rappresentare un'unica fonte di cellule per sostituire il tessuto danneggiato.
Questo tipo di trattamento richiede molto tempo e lavoro per identificare la mutazione genetica di ogni persona affetta da EB, sviluppare un approccio specifico di editing del DNA, coltivare le cellule per settimane e poi eseguire un intervento chirurgico clinico per effettuare gli innesti cutanei. Richiede inoltre una ricerca molto specializzata e strutture per la coltura cellulare. Nonostante questi ostacoli, sono stati riportati importanti progressi che dimostrano che questo processo può funzionare. La sezione successiva illustra uno di questi casi di successo.
Riflettori puntati sulla ricerca europea: una storia di successo
Nel 2017 un gruppo di ricercatori e clinici guidati dal Professor Michele De Luca ha riportatosuccesso nell'utilizzo della terapia genica e di innesti di pelle coltivata in laboratorio per salvare un ragazzo che presentava una mancanza di oltre l'80% della sua pelle. Il bambino soffriva di JEB causata da mutazioni del gene della laminina (LAMB3) ereditato da entrambi i genitori. Tutti i trattamenti, compresi diversi approcci estremi, avevano fallito. I medici ritenevano che il bambino avesse poche possibilità di sopravvivenza. Le autorità governative hanno concesso l'autorizzazione all'uso compassionevole di una terapia genica sperimentale che era stata testata in precedenza solo in due casi clinici, ciascuno dei quali riguardava un solo paziente. Anche i genitori del bambino hanno acconsentito a provare la procedura, pur essendo stati informati che il bambino avrebbe potuto non sopravvivere all'intervento.
I ricercatori hanno iniziato raccogliendo campioni di pelle dal bambino e coltivando queste cellule in laboratorio per ottenere gruppi di cellule contenenti cellule staminali epidermiche (della pelle). È stato quindi utilizzato un vettore virale per aggiungere una copia funzionale del gene LAMB3 al DNA delle cellule staminali epidermiche. Questo processo non ha “corretto” la mutazione originale nel DNA del ragazzo, ma ha aggiunto una nuova copia del gene LAMB3 ai cheratinociti del ragazzo. Questo nuovo gene consente alle cellule di produrre proteine laminine funzionali. Le cellule con il gene LAMB3 funzionale aggiunto sono state quindi utilizzate per coltivare fogli di epidermide in laboratorio utilizzando metodi di coltura avanzati. Questo processo ha richiesto diverse settimane. I piccoli pezzi di pelle coltivati in laboratorio sono stati poi innestati sul ragazzo in due procedure separate a distanza di un mese l'una dall'altra.
Il successo di questo trattamento è molto incoraggiante; alcuni potrebbero persino definire il risultato miracoloso, poiché ha superato le aspettative.
Purtroppo, trattamenti di terapia genica come questi non sono ancora approvati clinicamente né ampiamente disponibili. La medicina personalizzata come questa è costosa e richiede strutture speciali e il lavoro di molte persone. Si spera che trattamenti come questo diventino clinicamente approvati, accessibili e ampiamente disponibili per molte forme di EB.
Il futuro è promettente. Il successo del trattamento di questo ragazzo, così come i risultati positivi di altri studi, ha portato a diversi nuovi studi clinici. M. De Luca del Centro di Medicina Rigenerativa “Stefano Ferrari” di Modena, Italia, insieme a JW Bauer dell'Ospedale Universitario di Dermatologia di Salisburgo, Austria, stanno conducendo diversi studi clinici che utilizzano procedure simili ma più avanzate per sostituire le mutazioni nei geni COL7A1 e COL17A1 (Studio 1, Studio 2). Anche ricercatori di altre istituzioni stanno sviluppando le proprie terapie geniche, come la sperimentazione clinica guidata da Jean Yuh Tang della Stanford University School of Medicine, che mira anch'essa alle mutazioni del gene COL7A1 alla base della DEB.
Altri approcci terapeutici
La creazione di pelle modificata con il DNA presenta sfide importanti: il processo è complesso, costoso e rischioso. Inoltre, potrebbero volerci anni prima che questo approccio diventi un trattamento realistico.
Queste sfide hanno spinto i ricercatori a pensare ad altri approcci per il trattamento dell'EB che potrebbero essere realizzati più rapidamente, approvati clinicamente e accessibili dal punto di vista finanziario alle persone affette da EB. Questi approcci coinvolgono cellule staminali, farmaci, estratti vegetali e molti altri trattamenti. I due riportati di seguito sono solo alcuni esempi.
Sono in fase di sviluppo creme e lozioni terapeutiche topiche per contrastare i problemi causati da mutazioni specifiche dell'EB. Un trattamento topico, Vyjuvek, è già stato approvato per l'uso nell'UE. Uno studio clinico di particolare interesse sta utilizzando una crema che cerca di riparare il gene COL7A1, che aiuterebbe le persone affette da EB distrofica. La crema contiene componenti che inducono le cellule della pelle a bypassare la mutazione del gene COL7A1 durante la produzione della proteina collagene VII.
I trapianti di midollo osseo sono utilizzati per trattare la leucemia e altri disturbi del sangue e del sistema immunitario. Tuttavia, alcuni gruppi di ricerca stanno studiando il trattamento dell'EB con trapianti allogenici di midollo osseo, in cui le cellule staminali del sangue provengono da donatori sani. Il vantaggio di questo approccio è che i trapianti di midollo osseo sono stati utilizzati con successo per molti anni e la tecnica è stata notevolmente migliorata nel corso del tempo. I risultati preliminari suggeriscono che le cellule staminali sane del midollo osseo (o le cellule prodotte da queste cellule staminali) possono produrre proteine del tessuto connettivo che mancano nella pelle, come il collagene VII nei soggetti affetti da DEB. Tuttavia, questo approccio presenta anche dei limiti, tra cui la necessità di un donatore immunocompatibile e, attualmente, i rischi associati sono elevati.
Un elenco più completo delle sperimentazioni cliniche sull'EB è disponibile all'indirizzo clinicaltrials.gov. (Si prega di notare che questo sito web pubblico elenca solo gli studi clinici. Non verifica se gli studi clinici elencati siano sicuri, scientificamente validi o condotti da istituzioni affidabili. Si prega di leggere le dichiarazioni di non responsabilità, di rivolgersi a operatori sanitari di fiducia e di informarsi sui rischi e sui potenziali benefici).
Per saperne di più
Risorse di DEBRA, una rete di sostegno e difesa dei pazienti con EB (in englese):
EB Research Network: un'associazione di beneficenza che sostiene la ricerca sull'EB, guidata da DEBRA Austria.