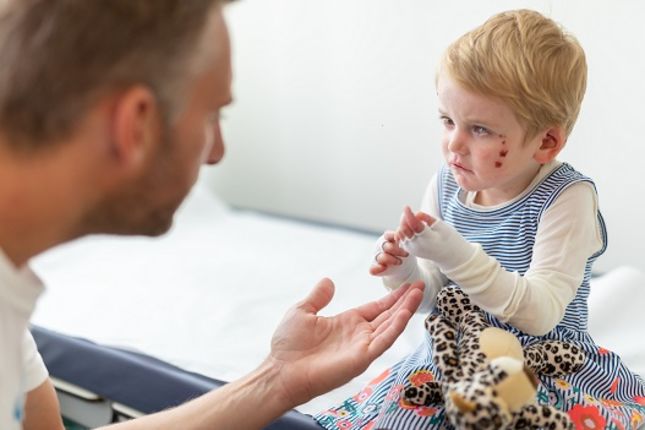
Epidermolysis Bullosa (EB) is a genetic disorder that disrupts the way cells and tissues are held together. There are different types of EB, but a common symptom of all EB is extremely fragile skin that easily tears or develops sores and blisters from even light friction or minor injury. There is currently no cure for EB, but gene therapy is already being used to treat people with EB, and promising research and clinical trials are exploring new ways that gene and cell therapy can help.
What do we know?
All forms of EB are caused by mutations in genes which affect how cells and tissues connect together. EB Simplex, Junctional EB, Dystrophic EB, and Kindler EB are the four main types of EB.
EB is relatively rare, estimated to affect 1 in 50,000 people.
EB visibly affects the skin, but this disorder can also cause complications in other organs, such as blisters and sores in the throat, upper airway and urinary tract. It may result in progressive systemic disease with life-limiting consequences, and may also lead to aggressive skin cancers.
There is currently no cure for EB, but a topical gene therapy has recently been approved to help treat wounds caused by EB.
What are researchers investigating?
There are many new devices, drugs and biological treatments being examined in clinical trials. Many of these studies aim to improve the management of EB. Some studies aim to ‘fix’ the underlying genetic mutations causing EB.
The development of new gene therapies may lead to the ability to restore healthy genes in skin cells. Working with epidermal (skin) stem cells, optimising techniques for growing skin in the lab, and improving tissue-grafting methods are other key components in developing successful gene therapies for EB.
What are the challenges?
Treating a genetic disorder is very challenging, because the cause lies in a person’s DNA. Treatments to compensate for gene mutations must repair the mutant gene or add a new functional copy of the gene to cells. This is not a simple task. Modifying cell DNA or adding new genes into cells carries potential risks, such as the possibility of cancer. Cell and skin transplants might carry risks such as bleeding, infection, and graft rejection. Careful clinical research aims to make sure that new treatments for EB are safe and effective.
A remaining challenge is also to make complex treatments, like gene therapies, affordable and accessible to everyone.
About Epidermolysis Bullosa
Epidermolysis Bullosa (EB) is a group of rare genetic disorders that cause very fragile, blistering skin. The blisters can be caused by friction to the skin, minor skin injuries and everyday activities, such as rubbing and scratching. These blisters can become painful sores and open wounds. In severe forms of EB, blisters and sores also develop in other tissues, such as in the mouth, throat, stomach, upper respiratory tract, and urinary tract.
There is a wide range of severity of EB. Mild types of EB may result in dealing with minor blisters throughout life, and others only have severe EB symptoms as children which improve with age. Some forms of EB can be life-threatening due to losing large amounts of skin. Others can cause extensive scarring, fusion of fingers and toes as well as other life-limiting complications. Some forms of the disease may lead to the development of skin cancer, reducing life expectancy
EB is rare, which makes estimating the total number of people who have EB challenging. DEBRA, a charity dedicated to EB, estimates that about 500,000 people worldwide have some form of EB.
All inherited types of EB are caused by mutations in a person’s DNA. These mutations disrupt one of several genes important for proteins that connect cells and tissue together. That is why EB is also referred to as a “connective tissue” disorder. Currently there are 16 genes in our DNA that have been linked to classical inherited EB.
How is EB inherited?
A gene mutation linked to EB can be passed down from parents to children. Our DNA contains two copies of every gene (except for genes on the X and Y chromosomes). Several forms of EB only require inheriting one copy of an EB-linked gene mutation. This is referred to as a “dominant” form of EB. The parent that passes on a dominant form of EB-linked gene mutation also has EB and its symptoms. Other forms of EB require inheriting an EB-linked gene mutation from both parents. These are called “recessive” forms of EB. Recessive forms of EB are often a surprise to a family because neither parent has the symptoms of EB and may not know they carry the mutation.
About the skin
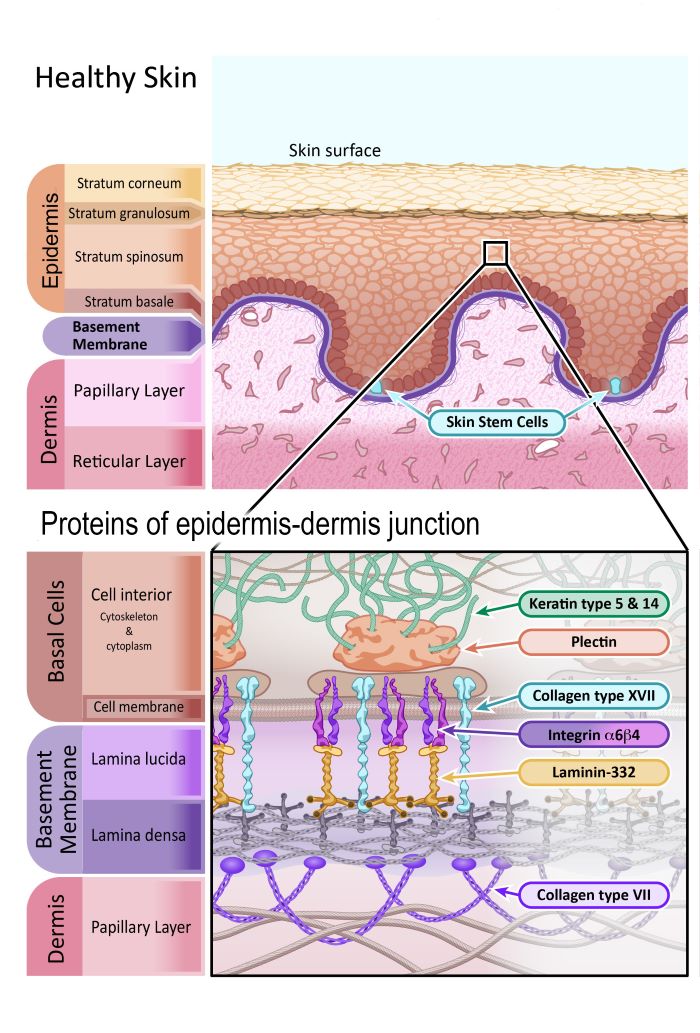
Our skin is a large and complex organ. It acts as a durable, flexible waterproof shield that keeps water in and bacteria out. It protects us from wind and weather, and helps us regulate our body temperature. It is where vitamin D is made in the body. Our skin has a huge number of specialised cells that allow us to sense temperature, textures, pressure and pain.
The two main layers of the skin are the epidermis (the outer layer) and dermis. Researchers further subdivide these layers into specific layers (or strata) of cells. You can see these sub-layers in the diagram below.
The epidermis is made up of mostly cells called keratinocytes. They are named keratinocytes because of the tough protein they produce called keratin – which is the same protein that makes up our hair and fingernails. Keratinocytes are made by skin stem cells found in the Stratum Basale sub-layer of the epidermis, and in hair follicles of the dermis. Skin stem cells are constantly making new keratinocytes to replace the skin cells that are continually falling or rubbing off. This makes skin stem cells very important for maintaining the health of our skin.
The dermis is the layer of skin that contains blood vessels, sensory cells for touch, temperature and pain, sweat glands, hair follicles and more. There are many different types of cells in the dermis, but most of the dermis is actually made of a flexible mesh of connective fibres and proteins. This mesh of proteins supports the structure of the skin and holds everything in place. The proteins involved in this mesh are mainly collagen and elastin.
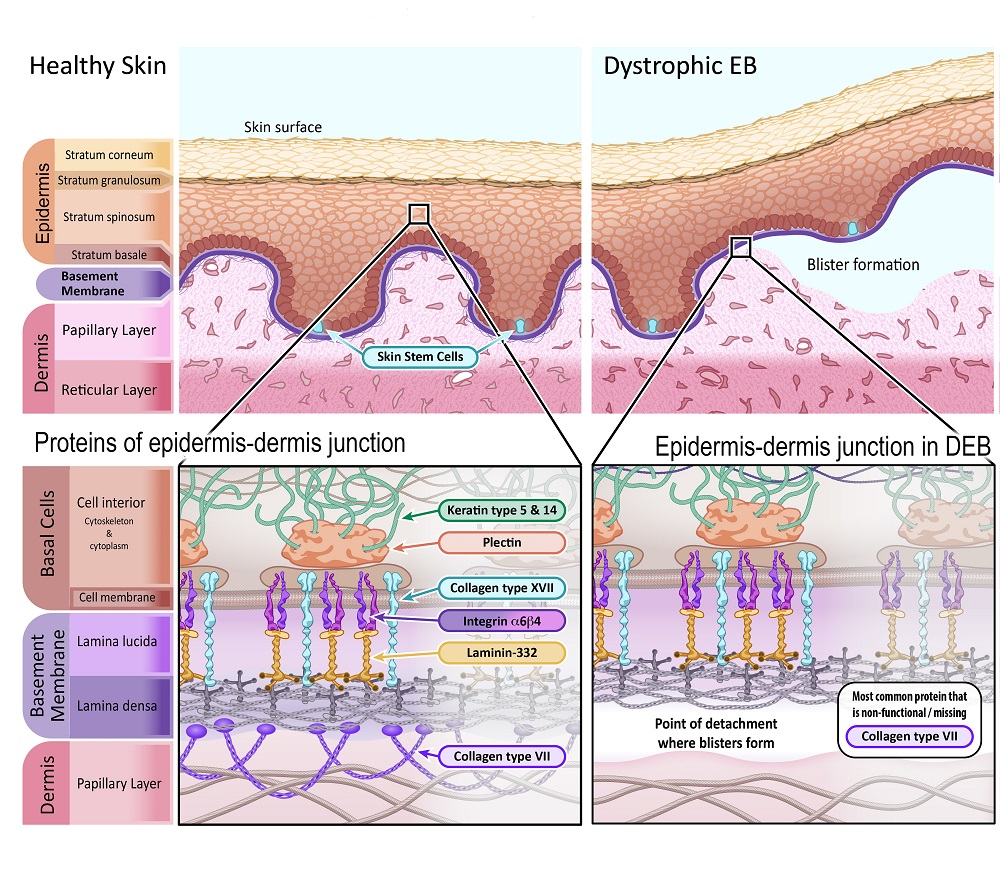
The connecting point between the epidermis and dermis is also important, particularly when discussing EB. The ‘basement membrane’ is a thin sheet of connective tissue important for binding the epidermis and dermis together. The basement membrane is primarily made of two types of proteins, laminin and collagen.
There are four main types of EB: EB Simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB) and Kindler EB (KEB). These categories are commonly distinguished by the layer of skin where blisters form. Although all forms of EB have blisters as a symptom, they are not caused by the same problem. The gene affected by an EB-linked genetic mutation dictates where, why and how extensively blistering and skin detachment occurs. It also determines what type of EB a person has.
Currently, there are 16 specific genes in our DNA that have been linked to classical EB. Mutations in these genes stop cells from making important functional proteins that hold the epidermis and dermis together. Knowing what role proteins normally have in holding skin together is critical to understanding the different types of EB. It also reveals why some types of EB are more severe than others, and why a treatment for one type of EB may not work for all.
Below are summaries that highlight the genes, their proteins and some specifics of where and why detachment occurs for the main forms of EB.
EB Simplex
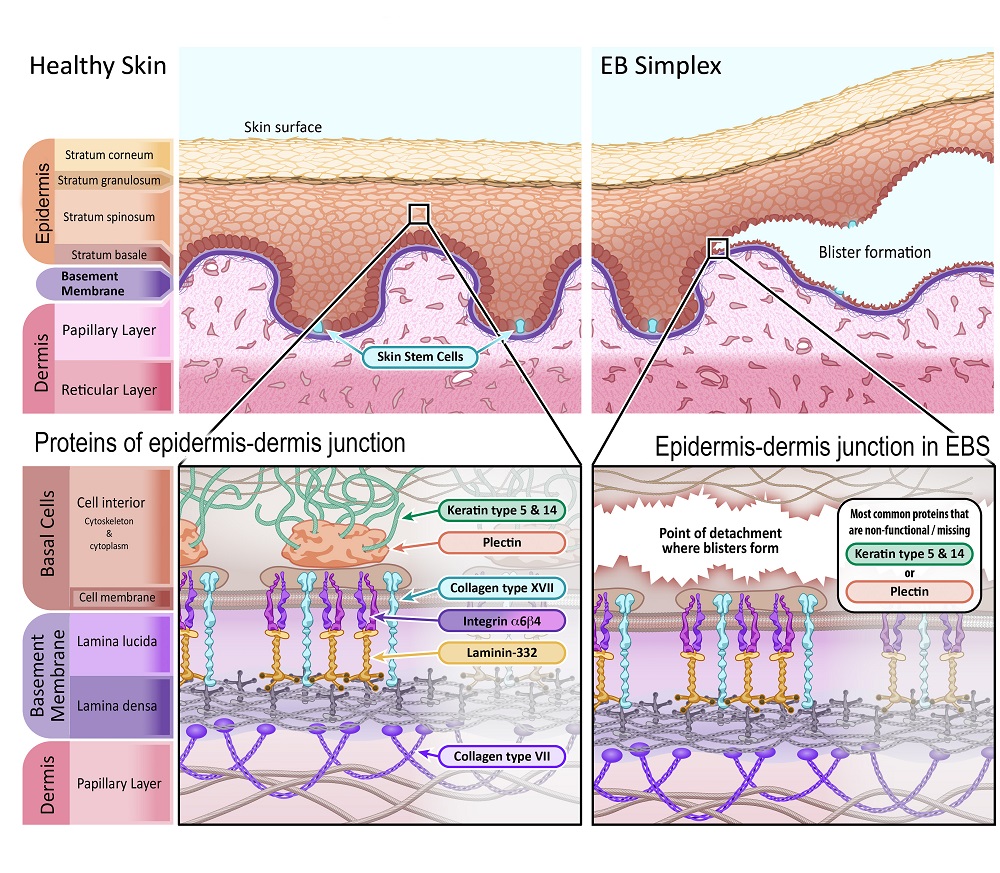
EB Simplex is the most common form of EB, making up about 70% of all EB cases. The severity of EBS symptoms can vary, ranging from skin fragility and minor blistering on hands and feet, to cases where blistering occurs all over the body, to more severe subtypes. EBS is predominantly inherited from parents as a dominant trait, meaning that only one parent needs to pass on the EB-linked gene mutation. That parent has EBS and likely has (or had) the associated symptoms.
In EBS, separation of the epidermis and dermis occurs within the basal cells in the epidermis (stratum basale), just above the basement membrane. The problem is that the proteins securing the basal cells to the basement membrane do not properly attach to the structural support within the cell (the cytoskeleton). This causes the basal cells to tear and rupture, leaving the bottom part of these cells attached to the basement membrane.
The primary proteins affected by gene mutations in EBS are:
Keratin type 5 – [Gene name:KRT5]
Keratin type 14 – [Gene name: KRT14]
Plectin – [Gene name: PLEC1]
Illustration 2: Diagram depicting healthy skin in comparison with skin affected by Epidermolysis Bullosa Simplex.
Junctional EB
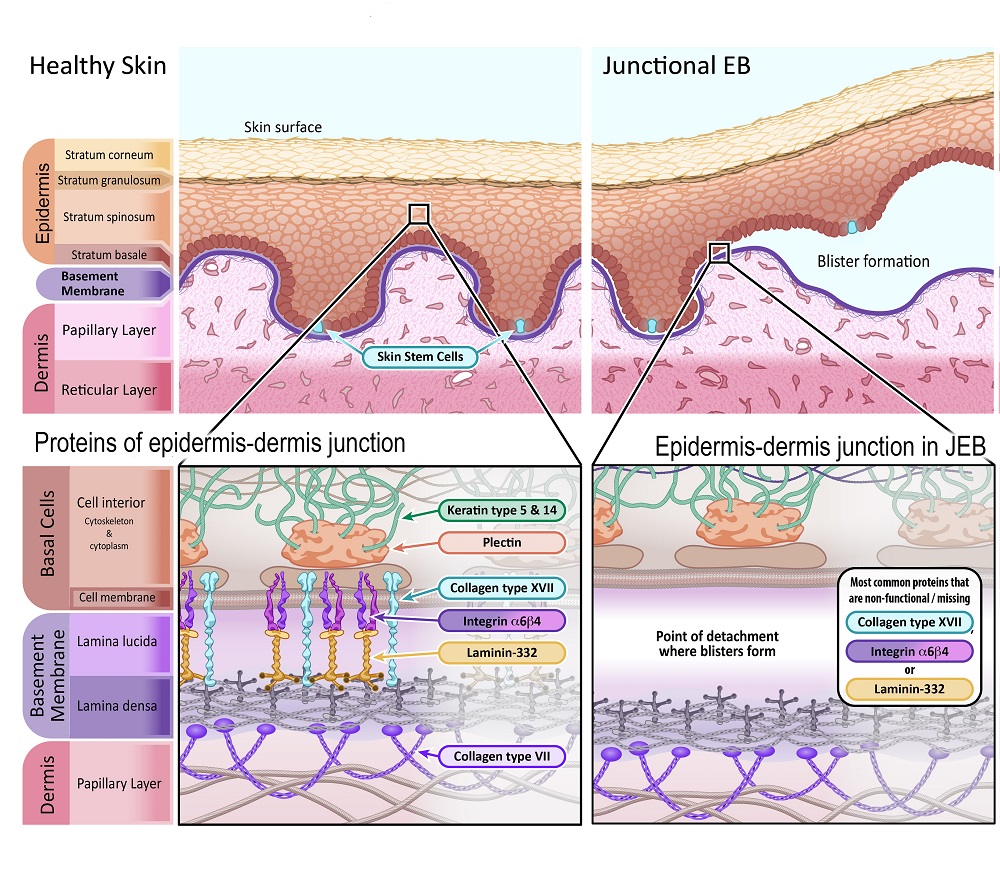
Junctional EB (JEB) makes up about 10% of all EB cases. JEB is inherited from parents as a recessive trait, meaning that an individual must have inherited an EB-linked gene mutation from both parents. Those parents likely did not have any symptoms of EB.
Some people affected by JEB will have “severe JEB” (previously called JEB generalised severe or Herlitz JEB). Blistering is all over the body, including the mouth, nose and throat. This blistering prevents newborn children from eating and breathing properly, causing malnutrition and lung failure. Tragically, this extreme subtype of JEB is life-threatening, and very few children with severe JEB live beyond two years old.
The other major subtype is “intermediate JEB ” (previously called JEB generalised intermediate or non-Herlitz JEB). Symptoms for this group are less serious, but still intense, and unfortunately, fatalities are still common. Individuals with intermediate JEB can live to adulthood, but often have large amounts of scarring, problems with nails and teeth, as well as other life-limiting complications.
In JEB, separation of the epidermis and dermis occurs at the basement membrane, specifically at the lamina lucida sublayer. This sublayer is made of proteins that anchor the basal cells of the epidermis to the dermis. When proteins in this layer are missing or not functional, this junction point of the basement membrane doesn’t hold together.
The primary proteins affected by gene mutations in JEB are:
Collagen type XVII (also known as BPAG2) – [Gene name: COL17A1]
Laminin-332 (also known as Laminin-5) – [Gene names: LAMA3, LAMB3 and LAMC2]
Illustration 3: Diagram depicting healthy skin in comparison with skin affected by Junctional Epidermolysis Bullosa.
Dystrophic EB
Dystrophic EB (DEB) makes up about 20% of all EB cases. DEB can be inherited as both a dominant or recessive trait, depending on the specific gene mutation.
The severity of symptoms greatly varies for DEB. In many cases, the dominant form of DEB causes mild blistering and symptoms, which lets these individuals lead a life with mild impairment. Most individuals with the recessive DEB type have to face many more limitations, as blistering and scarring is far more severe, and there is an increased likelihood of skin cancer. Additional symptoms may include disfigurement as blisters and wounds heal; fingers and toes fusing together, limited joint movement, and narrowing of the oesophagus (food pipe), making it difficult to swallow.
In DEB, separation of the epidermis and dermis occurs under the basement membrane. The primary protein that binds the basement membrane to the dermis is collagen VII. This protein forms anchoring fibrils that weave into the structural proteins of the dermis. Without collagen VII, the basement membrane easily detaches from the dermis.
The primary protein affected by a gene mutation in DEB is:
Collagen type VII – [Gene name: COL7A1]
Illustration 4: Diagram depicting healthy skin in comparison with skin affected by Dystrophic Epidermolysis Bullosa.
Kindler EB
Kindler EB (KEB) is a rare subtype of inherited EB. KEB is a recessive form of EB, meaning that an individual inherits the gene mutation from both parents. Symptoms can range from mild to severe, and include skin fragility, sensitivity to light, discolouration and thickening of the skin. Gum disease, mouth blisters and inflamed intestines may affect eating, and those with KEB have an increased risk of developing skin cancer.
KEB can affect different and multiple skin layers. When the affected protein is missing or not functional, it disrupts the ability of keratinocytes (skin cells) to grow and divide normally, and to attach the epidermis to the dermis.
The primary protein affected by a gene mutation in KEB is:
Kindlin-1 – [Gene name: FERMT1]
Our partners at the EB Research Network have information on EB and its classification
Treatments for EB primarily focus on preventing or relieving the symptoms. Preventative measures may involve using wrappings and padding to reduce friction, bumps and minor trauma to the skin. Treatments may also aim to prevent infections and to help blisters and sores heal.
In more severe cases of EB, medications may be used to prevent infections of open wounds and sores. Surgery may also be carried out to prevent fingers and toes from fusing or the throat and oesophagus (food pipe) from becoming too narrow.
In extreme cases of EB, doctors may cover large open wounds by conducting skin grafts. However, grafting skin from donors is very rare because the immune system will almost always reject donor skin grafts, even if a patient and donor are closely matched. Therefore, doctors often use temporary skin substitutes to cover large open wounds. New ‘biological skin substitutes’ are also becoming more common. These advanced skin substitutes aim to improve wound healing by using synthetic meshes that cells like to grow on, proteins found in the skin and sometimes even living keratinocytes.
Gene Therapy for Dystrophic EB
There is currently one gene therapy approved in the EU which targets the underlying genetic cause of dystrophic EB. Vyjuvek is a topical gene therapy, which means it is applied directly to the skin in the form of a gel. It is used to help heal the wounds and lesions that occur because of EB.
Vyjuvek works by delivering two healthy copies of the COL7A1 gene to keratinocytes and fibroblasts in the skin, allowing them to produce collagen and aiding wound healing. Vyjuvek delivers the COL7A1 gene into cells using an inactivated form of the herpes simplex virus (HSV-1) as a vector. HSV-1 is the same virus which causes cold sores. However, these virus particles have been engineered to prevent them from replicating, which makes them safe to use and means they do not cause infection.
HSV-1 cannot pass through the skin barrier, so this treatment can only be applied to open wounds or lesions. This means it can only help wounds heal, and can’t prevent them from developing. The treatment is not permanent, so ongoing application over time is required to maintain its clinical effectiveness.
Current research
Improving quality of life
There are many researchers, drug companies and medical supply companies working to develop better products to improve managing the symptoms of EB.
Products being examined range from better bandages and wraps to ointments and lotions that help the skin heal.
Making 'New' Skin
The ideal way to fix a genetic disorder is to correct or replace a mutated gene in a person’s DNA. Researchers are creating gene therapies that do just this. New discoveries and technologies, such as CRISPR-Cas9 gene editing, are greatly speeding up how DNA and genes can be edited in cells. However, changing the DNA in a person’s cells is not a simple task. The process might also carry serious risks. If adding or editing a gene goes wrong it can cause cancer, add new complications or worsen an individual’s condition. Clinical trials are important for identifying risks of new treatments and prove that they really work.
In many cases, stem cells go hand-in-hand with gene therapies. For EB gene therapies, researchers want to edit the DNA of skin stem cells, rather than other types of skin cells. This is because adding (or editing) a gene in skin stem cells will cause that gene to be passed on to every other cell the stem cells make. This is important because almost all keratinocytes in our skin are replaced every 4-5 weeks. Additionally, skin stem cells continually renew themselves, so any DNA edits introduced to a stem cell could potentially be ‘permanent’ (or last at least as long as the stem cells survive).
Several clinical trials are testing methods of editing the DNA of EB patient skin stem cells to grow ‘new’ healthy skin for grafting. Although the methods and details of these different clinical trials vary, the overall idea and process is generally the same. Skin samples are taken from a person affected by EB and cultured in a lab; hopefully, these samples contain skin stem cells. DNA editing methods are then used to add or edit genes in the cells and restore normal protein production. Any skin stem cells with the new DNA-edits are then used in advanced cell culturing methods to grow layers of skin in the lab. Then, these lab-grown layers of skin can be grafted on to the patient. S
Some of these approaches involve using induced pluripotent stem cells (iPSC), a type of stem cell that can be generated from cells in our body. These cells can propagate indefinitely and can give rise to every other cell type in the skin. They could represent a single source of cells to replace damaged tissue.
This kind of treatment requires lots of time and work to identify the genetic mutation for each person affected by EB, develop a specific DNA-editing approach, growing cells over weeks and then clinical surgery to conduct the skin grafts. It also requires very specialised research and cell-growing facilities. Despite these obstacles, there have been major advances reported that show this process can work. The next section discusses one such success story.
Spotlight On European Research: A Success Story
In 2017 a group of researchers and clinicians lead by Professor Michele De Luca reported successfully using gene therapy and lab-grown skin grafts to save a boy that was missing over 80% of his skin. The boy was suffering from JEB caused by mutations in the laminin (LAMB3) gene he inherited from both parents. All treatments, including several extreme approaches, had failed. Clinicians thought the boy was unlikely to survive. Government authorities granted permission for “compassionate use” of a preliminary gene therapy treatment that had only been trialed previously in two case studies that looked at only one patient, respectively. The boy’s parents also agreed to try the procedure, even after being told that the boy might not survive the procedure itself.
Researchers began by collecting skin samples from the boy and growing these cells in the laboratory to obtain clusters of cells that contain epidermal (skin) stem cells. A viral vector was then used to add a functional copy of the LAMB3 gene to the DNA of the epidermal stem cells. This process did not 'correct' the orginal mutation in the boy's DNA - it added a new copy of the LAMB3 gene to the boy’s keratinocytes. This new gene allows cells to make laminin proteins that are functional. The cells with the added functional LAMB3 gene were then used to grow sheets of epidermis in the laboratory using advanced culturing methods. This process took multiple weeks. The small pieces of lab-grown skin were then grafted on to the boy in two separate procedures one month apart.
The boy survived, and is now living with skin made from gene-edited cells over 80% of his body. The researchers examined this skin and say it looks remarkably like normal skin in many ways.
The success of this treatment is very exciting; some may even say the outcome was miraculous because it exceeded expectations. Unfortunately, gene therapy treatments like these are not yet clinically approved or widely available. Personalised medicine like this is expensive, requiring special facilities and the work of many people. Hopefully treatments like this will become clinically approved, affordable and widely available for many forms of EB.
The future is promising. The success of this boy's treatment, as well as positive results in other studies, has led to several new clinical studies. M. De Luca from the Centre for Regenerative Medicine “Stefano Ferrari” in Modena, Italy, together with JW Bauer from the University Hospital of Dermatology in Salzburg, Austria are leading several clinical studies using similar but more advanced procedures to replace mutations in the COL7A1 and COL17A1 genes (Study 1, Study 2). Researchers at other institutions are also developing their own gene therapies, such as the clinical trial lead by Jean Yuh Tang from Stanford University School of Medicine, which is also targeting DEB underlying mutations in the COL7A1 gene.
Other Therapeutic Approaches
As stated in the previous section, making DNA-edited skin has major challenges; the process is complex, expensive and risky. Also, it may be years before this approach becomes a realistic treatment.
These challenges have prompted researchers to think of other approaches to treating EB that might be more quickly realised, clinically approved and financially accessible to those affected by EB. These involve stem cells, drugs, plant extracts and many other treatments. The two below are just examples.
Therapeutic topical creams and lotions are being developed to counter the problems of specific EB mutations. One topical treatment, Vyjuvek, has already been approved for use in the EU. A clinical study of particular interest is using a cream that tries to fix the COL7A1 gene, which would help individuals with dystrophic EB. The cream contains components that promote skin cells to skip over the mutation in the COL7A1 gene when making the collagen VII protein.
Bone marrow transplants are used to treat leukaemia and other blood and immune system disorders. However, some research groups are exploring the treatment of EB with allogeneic bone marrow transplants, where blood stem cells come from healthy donors. The advantage of this approach is that bone marrow transplants have been successfully used for many years and the technique has been greatly improved over that time. Preliminary results suggest that healthy stem cells from bone marrow (or cells made by these stem cells) can make connective-tissue proteins that are missing in the skin, such as collagen VII in individuals with DEB. However, this approach also has its limitations including the requirement for a human immuno–matched donor, and currently the associated risks are high.
A more comprehensive list of clinical trials for EB can be found at clinicaltrials.gov. (Please note that this public website only lists clinical trials. It does not check if the listed clinical trials are safe, scientifically sound or carried out by reputable institutions. Please read any disclaimers, speak to trusted healthcare providers and learn about the risks and potential benefits.)
Find Out More
Resources from DEBRA, a network for EB patient advocacy and support:
EB Research Network: an alliance of charities supporting research in EB, spearheaded by DEBRA Austria