Advanced Therapy Medicinal Products (ATMPs)
According to the European Medicines Agency (EMA), ATMPs are medicines for human use that are based on genes, tissues or cells. They are medicines for human use with an active therapeutic substance based on at least one of the following: 1) technology to modify patient genome, 2) recombinant nucleic acid or genes, 3) substantially manipulated cells, 4) cells intended for a different essential function in the patient versus the donor, 5) engineered tissue.
They offer new opportunities for the treatment of diseases and injuries with unmet medical needs.
ATMPs are classified into three main categories:
- Gene Therapy Medicinal Products (GTMPs),
- Somatic cell therapy medicinal product (sCTMPs) and,
- Tissue-engineered products (TEPs).
In addition, combined ATMPs contain one or more medical devices as an integral part of the medicine. (See "Therapy Classification” on EuroGCT)
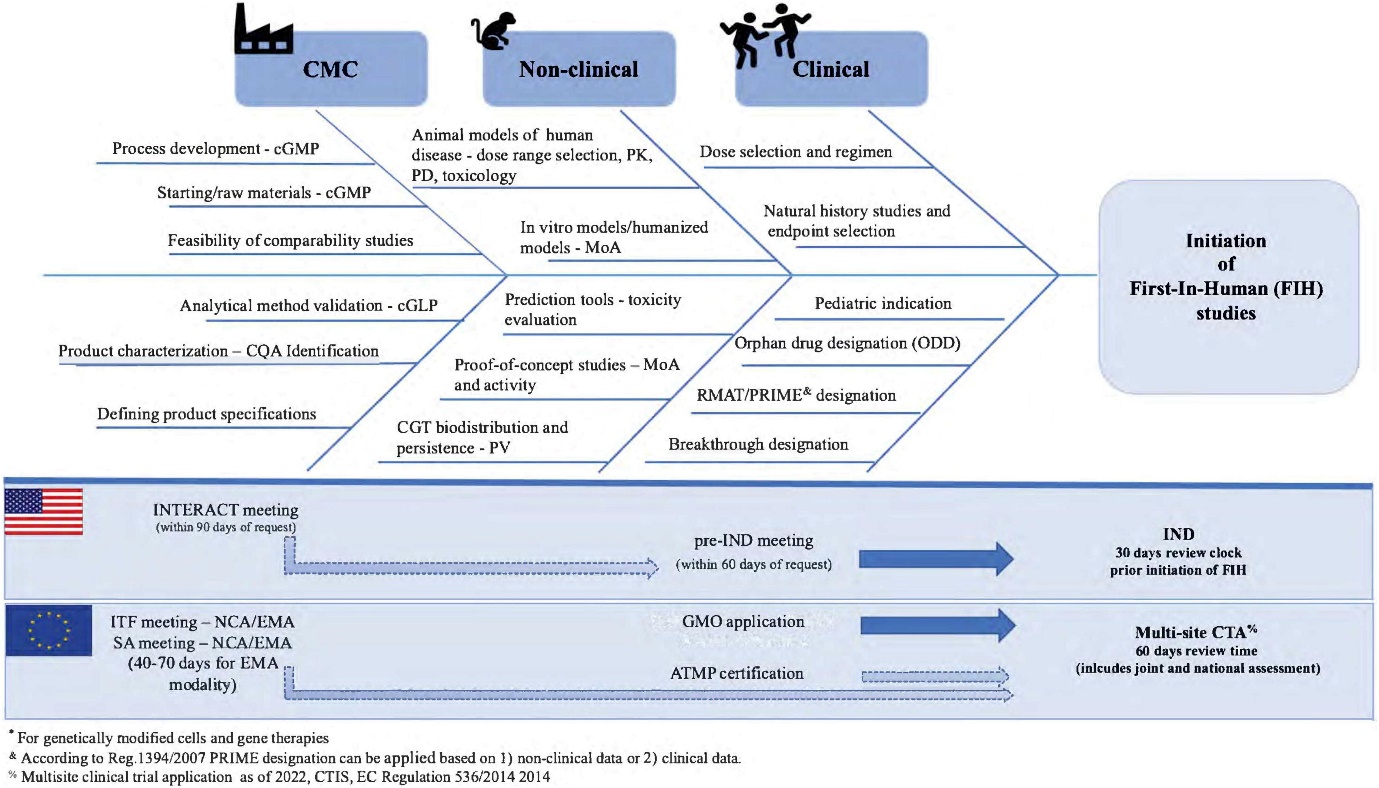
Before these therapies are offered to patients, the products must complete a development phase that adheres to the guidance of regulatory bodies such as the EMA. At the beginning of drug development, developers of Gene Therapy Medicinal Products (GTMPs) and Cell-Based Medicinal Products (CBMPs) need to navigate regulatory requirements related to non-clinical studies (See “Non-clinical Development Flowchart” on EuroGCT).
At this early stage, developers have to address key questions such as “what is the mechanism of action?”, “what is the efficacy?” and “what are the potential toxicological concerns?”. At later development stage, they also need to comply with various standards, including Good Laboratory Practice (GLP), Good Manufacturing Practice (GMP) (See “Good Manufacturing Practice for ATMPs” on EuroGCT), and Good Clinical Practice (GCP). (See “Clinical trials” on EuroGCT). The assessment of the quality, safety and efficacy of ATMPs is conducted by the EMA’s Committee for Advanced Therapies (CAT).
The CAT provides scientific recommendations on the classification of ATMPs (See “CAT scientific recommendations on classification” on EuroGCT) and is responsible for preparing a draft opinion on each ATMP marketing authorisation application submitted to the EMA. This draft is then reviewed by the Committee for Medicinal Products for Human Use (CHMP), which adopts the final opinion on the marketing authorization of the medicinal product (See “Market Access for ATMPs” on EuroGCT). The CAT also participates in procedures that deliver advice on the conduct of efficacy follow-up, pharmacovigilance or risk-management systems for ATMPs (See “Support for innovative medicines’ development at the EMA level” on EuroGCT).
More information can be found in Regulation (EC) 1394/2007 and in the CAT Work Plan 2025.
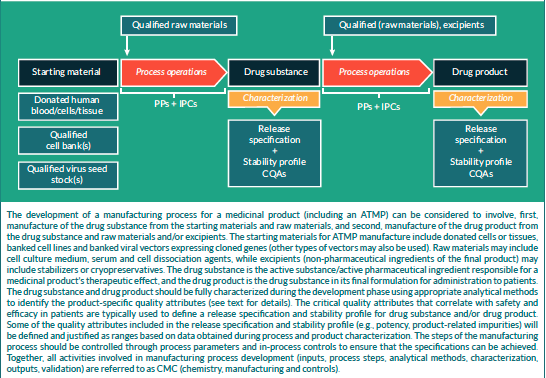
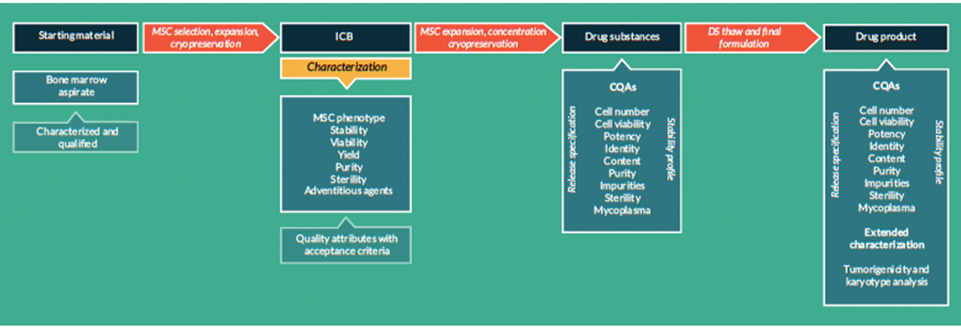
Manufacturing and control are key steps in drug development. They are involved in the production and the characterisation of the medicinal substance (also called drug substance or DS) which is then formulated to become the medicinal product (also called drug product or DP). This product is released according to well-defined criteria and it may be stored before being administered to patients. The terms “manufacturing, control and formulation” are collectively referred to as “CMC” which stands for “Chemistry, Manufacturing and Control”. CMC along with preclinical and clinical activities are necessary to transition a potential medicine from discovery phase to clinical trials. Clinical trial applications in Europe (See “Clinical trials” on EuroGCT) and Investigational new drug applications in the USA are reviewed by regulatory agencies and have to be cleared before initiating clinical trials. These applications must include information related to non-clinical pharmacology and toxicology studies, manufacturing and control, clinical protocols and investigator information. The figure 1 below titled “CMC in drug development” provides further details on this process. Guidance documents help developers in the rapidly evolving field of ATMPs in which scientific achievements often translate quickly into therapeutic innovations. The mapping of the European landscape and specificity of ATMPs guidance has been published in June 2025 by Aurélie Mahalatchimy et al.