Médicaments de Thérapie Innovante (MTI)
Selon l'Agence européenne des médicaments (EMA, de l’anglais European Medicines Agency), les Médicaments de Thérapie Innovante (MTI) sont des médicaments à usage humain basés sur des gènes, des tissus ou des cellules. Il s'agit de médicaments à usage humain dont la substance thérapeutique active repose sur au moins l'un des éléments suivants : 1) une technologie permettant de modifier le génome du patient, 2) des acides nucléiques ou des gènes recombinants, 3) des cellules ayant subi une manipulation substantielle, 4) des cellules destinées à remplir une fonction essentielle différente chez le patient par rapport au donneur, et 5) des tissus modifiés.
Ils offrent de nouvelles opportunités pour le traitement de maladies et de pathologies dont les besoins médicaux ne sont pas satisfaits.
Les MTI sont classés en trois catégories principales :
- les thérapies géniques (GTMPs, de l’anglais Gene Therapy Medicinal Products),
- les thérapies à base de cellules somatiques (sCTMPs, de l’anglais Somatic Cell Therapy Medicinal Products), et
- les médicaments issus de l'ingénierie tissulaire (TEPs, de l’anglais Tissue-Engineered Products).
En outre, les médicaments combinés contiennent un ou plusieurs dispositifs médicaux faisant partie intégrante du médicament. (Voir « Classification des thérapies » sur le site EuroGCT)
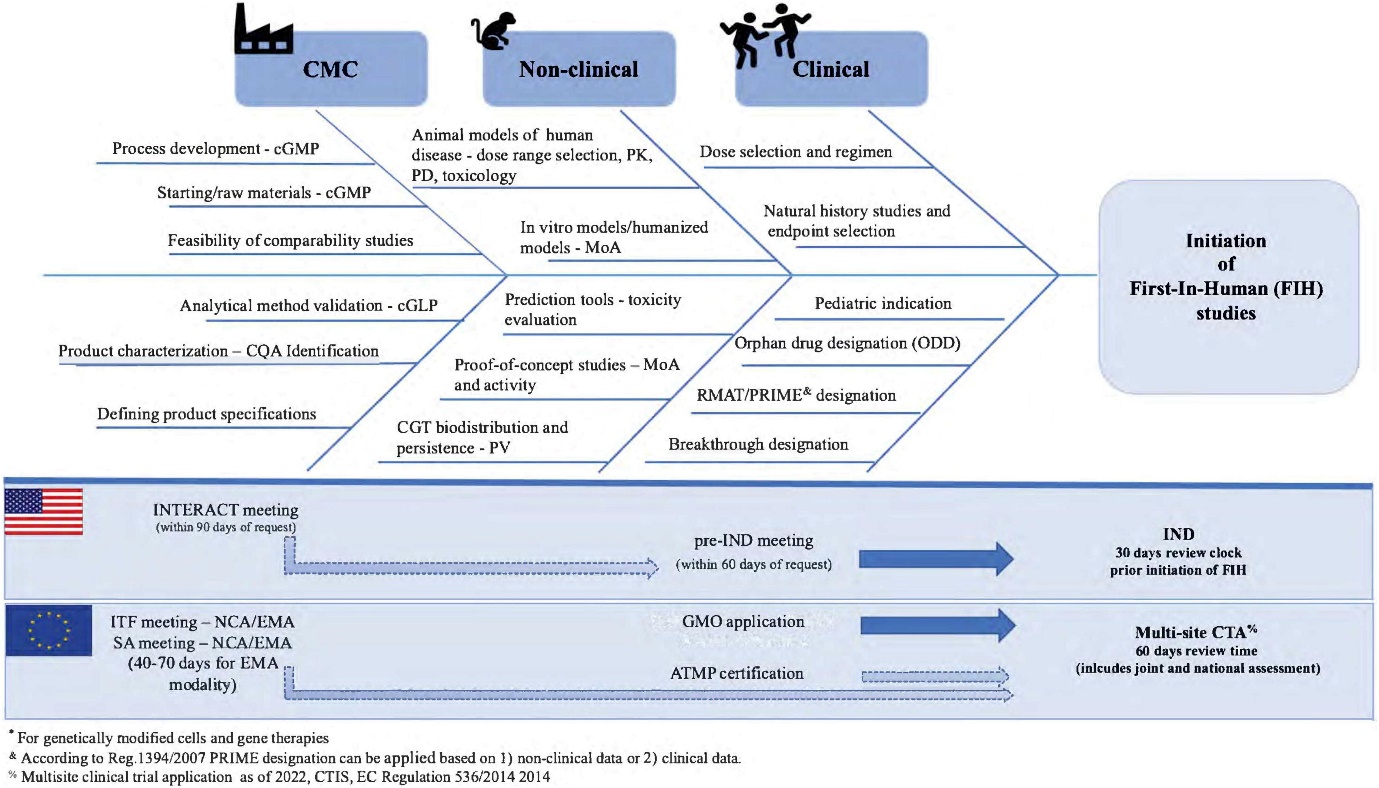
Avant que ces thérapies ne soient proposées aux patients, les produits doivent passer par une phase de développement qui respecte les lignes directrices d'organismes de réglementation tels que l'EMA. Au début du développement d'un médicament, les développeurs de GTMPs et de médicaments à base de cellules (CBMPs de l’anglais Cell-Based Medicinal Products) doivent se conformer aux exigences réglementaires relatives aux études non cliniques (voir « Non-clinical Development Flowchart » sur le site EuroGCT). À ce stade précoce, les développeurs doivent répondre à des questions clés telles que « quel est le mécanisme d'action ? », « quelle est l'efficacité ? » et « quels sont les risques toxicologiques potentiels ? ». À un stade de développement plus avancé ils doivent également respecter diverses normes, notamment les Bonnes Pratiques de Laboratoire (BPL), les Bonnes Pratiques de Fabrication (BPF) (Voir « Bonnes Pratiques de Fabrication des MTI » sur le site EuroGCT) et les Bonnes Pratiques Cliniques (BPC). L'évaluation de la qualité, de la sécurité et de l'efficacité des MTI est effectuée par le Comité des thérapies innovantes (CAT, de l’anglais Committee for Advanced Therapies) de l'EMA.
Le CAT fournit des recommandations scientifiques sur la classification des MTI (Voir « Recommandations scientifiques du CAT sur la classification ») et est chargé de préparer un projet d'avis sur chaque demande d’autorisation de mise sur le marché de MTI soumise à l'EMA. Ce projet est ensuite revu par le Comité des médicaments à usage humain (CHMP, de l’anglais Committee for Medicinal Products for Human Use), qui adopte l'avis final sur l'autorisation de mise sur le marché du médicament (Voir « Accès au marché pour les médicaments de thérapie innovante » sur le site EuroGCT). Le CAT participe également aux procédures qui délivrent des conseils sur la conduite du suivi de l'efficacité, de la pharmacovigilance ou des systèmes de gestion des risques pour les MTI (Voir « Soutien au développement de médicaments innovants au niveau de l'EMA » sur le site EuroGCT). Des informations complémentaires sont disponibles dans le Règlement (CE) N° 1394/2007 et dans le Programme de travail 2025 du CAT (en anglais).
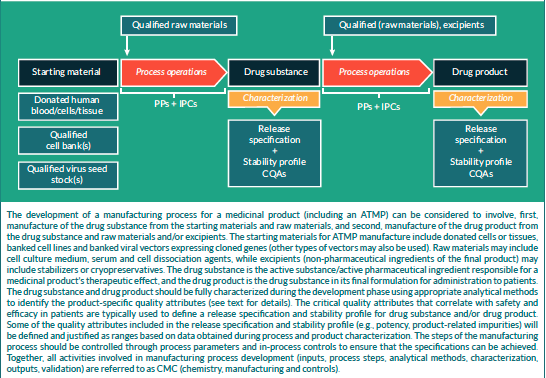
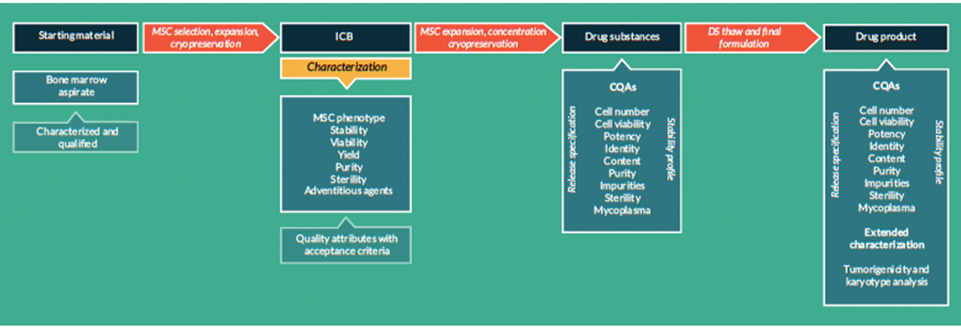
La fabrication et le contrôle sont des étapes clés du développement d'un médicament. Ils interviennent dans la production et la caractérisation de la substance médicamenteuse (également appelée substance médicament ou DS, de l’anglais Drug Substance) qui est ensuite formulée pour devenir le médicament (ou DP, de l’anglais Drug Product). Ce médicament est libéré selon des critères bien définis et peut être stocké avant d'être administré aux patients. Les termes « fabrication, contrôle et formulation » sont collectivement désignés par l'acronyme « CMC », de l’anglais « Chemistry, Manufacturing and Control » qui signifie « chimie, fabrication et contrôle ». La CMC ainsi que les activités précliniques et cliniques sont nécessaires pour faire passer un médicament potentiel de la phase de découverte aux essais cliniques. Les demandes d'essais cliniques en Europe (voir Clinical trials sur le site EuroGCT, en anglais) et les demandes de nouveaux médicaments expérimentaux aux États-Unis sont examinées par les agences de réglementation et doivent être approuvées avant le début des essais cliniques. Ces demandes doivent inclure des informations relatives aux études pharmacologiques et toxicologiques non cliniques, à la fabrication et au contrôle, aux protocoles cliniques et aux informations concernant l'investigateur. La figure 1 ci-dessous intitulée « CMC dans le développement des médicaments » fournit de plus amples détails sur ce processus (en anglais). Des documents d'orientation aident les développeurs dans le domaine des MTI, secteur en rapide évolution où les avancées scientifiques se traduisent souvent et rapidement par des innovations thérapeutiques. Voir notamment l’article Mapping of the European landscape and specificity of ATMPs guidance published in June 2025 par Aurélie Mahalatchimy et al.