Help and support
Help and support
- FAQ's
- Glossary
Medical applications of tissues and cells (T&C), whether from human or animal origin, can be various. While the legal distinction between transplants and Advanced Therapy Medicinal Products (ATMPs) has been widely commented, the legal classification of health technologies based on the association between medical devices (MD) and T&C is poorly explored and known. Yet, from their collection to the patient’s administration, they have to follow the appropriate regulatory pathways in accordance with their legal classification. First, the MD and the T&C have to comply with the respectively applicable European legislations: the regulations on MD and on substances of human origin for T&C of human origin. Second, when T&C are substantially manufactured into medicinal products, they would be categorised as biological medicinal products, most of the time as ATMPs. The association between T&C and MD may be external, and MD can be companion diagnostics to a transplant or an ATMP. Parallel regulatory pathways have to be followed by each product. But the association between T&C and medical devices may also be integrative. Depending on what is regulatorily considered as the dominant part in the overall product, the latter can be either a medicine or a MD.
On the one hand, if the T&C are the dominant part, the overall product should be a combined ATMP. This is a hybrid sub-category of ATMP that should encompass certain organoids. On the other hand, if the action of the T&C is only ancillary to that of the device, the overall product should be a MD. This latter type of products is not only defined in contrast to medicinal products, they are also following a completely different regulatory pathway to reach the market. While any ATMPs’ developer has to go through the centralised procedure to obtain a marketing authorisation (granted by the European Commission after evaluation by the European Medicines Agency), MDs’ manufacturers, on the other hand, have to “certify” their products. This requires the involvement of a Notified body – a non-state entity designated by a member state for the assessment of certain MDs – before commercialising MDs based on T&C. And while the European legislation distinguishes between gene therapy, cell therapy, tissue engineering, and combined ATMPs among ATMPs, it divides MDs into classes, depending on their purpose and their risk. Thus, their two distinct regulatory pathways are worst to be known, as each one has its own specific requirements. The distinction criteria between MDs and ATMPs are based on the viability of the tissues or cells, and primary or ancillary action of each part of the overall product. Yet, navigating through the various legal texts regarding definitions and scopes to identify the legal classification and applicable regulatory pathway(s) of health technologies may be challenging, especially as the association between MD and T&C adds another layer of complexity. This poster will clarify and discuss the regulatory pathways applicable to health technologies using T&C of human or animal origin, from the collection of the biological starting materials to the commercialisation of the final health technologies.
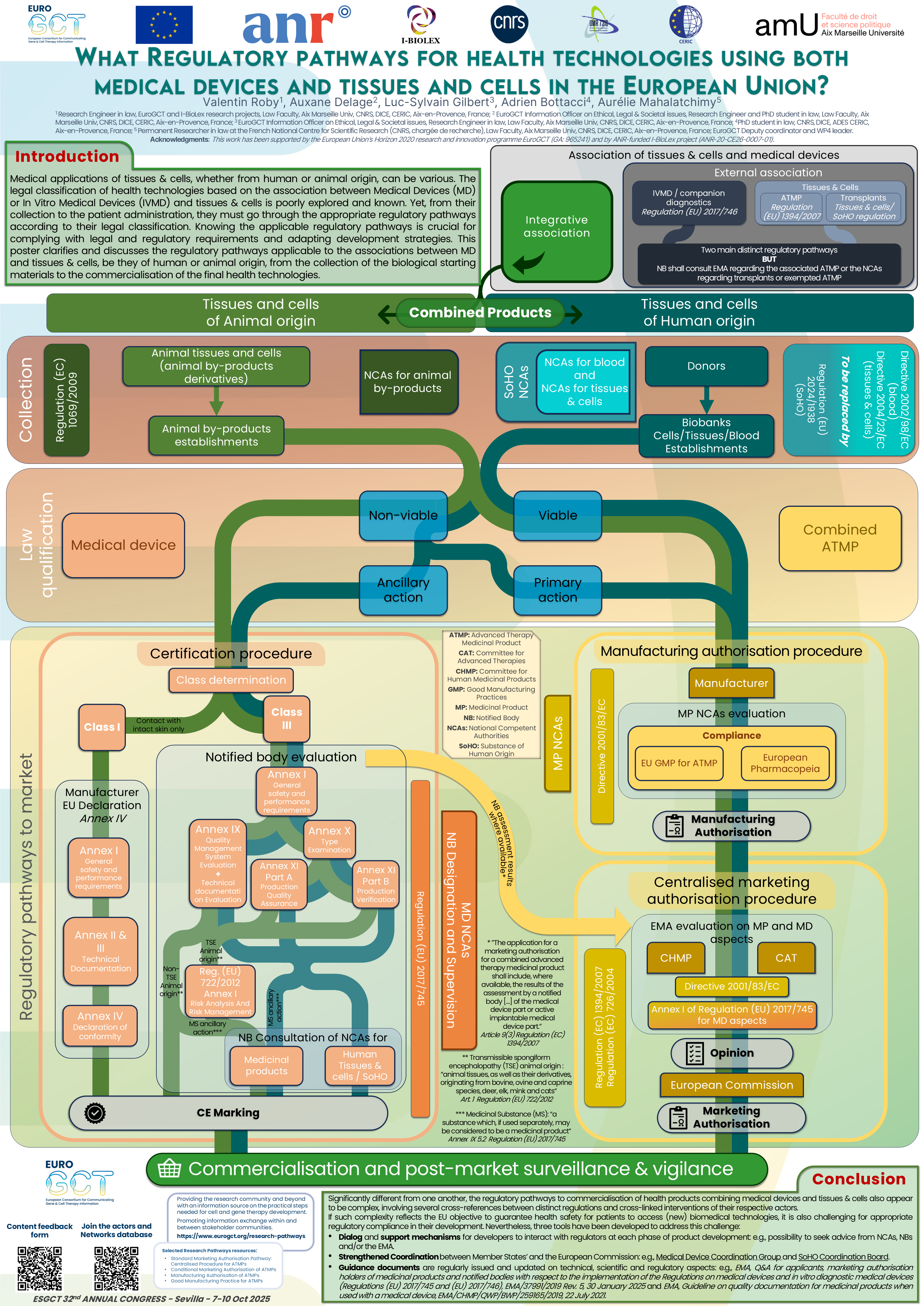
The pdf file of this poster can be downloaded from the attachments section at the bottom of this page
Valentin Roby, EuroGCT information officer on Ethical, Legal and Societal Issues
Auxane Delage, EuroGCT Information Officer on Ethical, Legal & Societal issues, Aix-en-Provence, France
Luc-Sylvain Gilbert, EuroGCT Information Officer on Ethical, Legal & Societal issues, Aix-en-Provence, France
Adrien Bottacci, PhD student in law, CNRS, DICE, ADES CERIC, Aix-en-Provence, France
Aurélie Mahalatchimy, EuroGCT Deputy coordinator and WP4 leader; UMR 7318 DICE CERIC, Aix-Marseille University, Centre National de la Recherche Scientifique, Marseille, Provence-Alpes-Côte d’Azur, France