1. ACT-EU: ACT-EU was created by a joint action from the EMA, the Heads of Medicines Agency (HMA) and the European Commission (DG SANTE and DG RTD). It was established in January 2022 with the impulse of the implementation of the new Clinical Trials Regulation. ACT-EU has a workplan with 11 priority areas aimed at enhancing the efficiency and effectiveness of clinical trials across the EU. The ACT-EU workplan 2025 – 2026 explains clearly the aim of this action is to accelerate Clinical Trials in the EU by supporting smarter clinical trials using multiple processes (innovation, regulatory and technological) to foster innovation. “The vision is to have better, faster and optimised clinical trials, benefitting patients and healthcare in Europe. Seamless coordination between stakeholders, regulators and ethics committees strengthens cross-border collaboration. The ACT-EU multi-stakeholder platform is central to this”.
ACT-EU translates this vision into a set of concrete projects that directly impact the planning and conduct of clinical trials. Priority areas include improving the use of CTIS, streamlining assessment procedures between NCAs and ethics committees, promoting innovative trial designs such as platform, adaptive or decentralised trials, and strengthening methodological and regulatory guidance for complex studies. The initiative also focuses on capacity building for regulators and stakeholders, enhanced transparency and use of real world data, and better integration of patient perspectives into trial design and governance. For sponsors and investigators, ACT-EU should progressively lead to more predictable timelines, clearer expectations across Member States and a more supportive environment for high quality, patient centred clinical research in the EU.
Find out more about ACT-EU on their website and also read the workplan for 2023-2026, and the latest yearly workplan for 2025-2026.
2. Clinical Trials Coordination Group (CTCG): The CTCG is a working group at the Heads of Medicines Agencies (HMA). It is composed of experts from national medicines agencies on the assessment and oversight of clinical trials. This group aims at enhancing the attractiveness of EU/EEA for clinical trials by promoting harmonization and optimization of regulations.
The CTCG develops common guidance, Q&A documents and best‑practice papers to support consistent implementation of the EU Clinical Trials Regulation across Member States. It works to align national approaches on key topics such as assessment of complex or multinational trials, use of CTIS, safety reporting, substantial modifications and transparency rules.
In addition, the CTCG serves as a forum where NCAs can discuss challenging or novel trial designs, share inspection and oversight experience, and coordinate positions on emerging scientific or regulatory issues. By doing so, it complements the work of EMA committees and working parties and provides sponsors with clearer, more predictable and harmonised regulatory expectations when planning and conducting multi‑country clinical trials in the EU/EEA.
Find out more about this group here. To read the CTCG mandate follow this link.
3. Pharmaceutical reform: The ongoing reform of the EU’s pharmaceutical legislation, commonly referred to as the “EU Pharmaceutical Package” including both the compromise text of a Regulation and the compromise text of a Directive, initiated in April 2023 and the subsequent political agreement reached on the 11 December 2025, is expected to introduce several regulatory changes that may affect the conduct of clinical trials involving ATMPs. One of the key objectives of the reform, as stated in Recitals 3 and 4 of the compromise text of a Regulation, is to strengthen the EU’s capacity to foster innovation while ensuring timely access to safe and effective medicines for patients across all Member States. The legislative proposals aim to simplify and modernise the regulatory environment governing pharmaceutical research, including clinical development processes. In particular, the reform seeks to reduce administrative burdens and streamline regulatory procedures, thereby making the EU a more attractive environment for clinical research.
The following proposals can be underlined:
a. While Regulation (EU) No 536/2014 provides a harmonised procedural framework for clinical trials, the compromise text of a Directive reshapes the incentives surrounding clinical development. In particular, Articles 81 and 82 of the compromise text of a Directive, read in light of Recital 60, establish a system of modulated regulatory data protection and market protection. These provisions allow for additional periods of protection where medicinal products address unmet medical needs or contribute to public health objectives. Although they do not directly regulate clinical trials, they create incentives for sponsors to generate robust clinical evidence, thereby indirectly influencing trial design, data validity criteria and development strategies.
b. Beyond these direct and incentive-based effects, the pharmaceutical reform significantly impacts the clinical trial ecosystem through the reorganisation of scientific evaluation and regulatory support mechanisms regarding:
- The restructuring of scientific committees, as reflected in Recital 37 of the compromise text of a Regulation, which addresses the integration of expertise previously concentrated within the Committee for Advanced Therapies into broader EMA structures such as the CHMP to contribute to a more streamlined and integrated scientific assessment. While this move does not directly regulate clinical trials, it affects the evaluation of clinical data generated during such trials, thereby influencing expectations regarding evidence generation, particularly for complex products such as ATMPs;
Scientific advice: The strengthening of EMA scientific advice plays a central role in supporting the marketing authorisation application of ATMP, including the shaping of their clinical development.
The EU Pharmaceutical Package establishes an incentive architecture linking clinical trial design to regulatory market protection. Under the compromise text of a Directive, a twelve-month prolongation of the regulatory market protection period is subject to whether the clinical trials supporting the marketing authorisation application use a relevant and evidence-based comparator in accordance with scientific advice provided by the EMA (Articles 81.2(b) and (c)), and/or whether the clinical trials evaluating the efficacy of the medicinal product and used for the marketing authorisation were conducted in more than one Member State (Articles 81.2(c) and (d)). Compliance, or justified non-compliance, with an EMA scientific advice has therefore tangible consequences for market exclusivity. Article 81.3 further mandates the EMA to develop scientific guidelines on comparator selection.
These incentives on comparative clinical evidence aim both to strengthen regulatory assessment and to generate data capable of supporting downstream health technology assessments and decisions on pricing and reimbursement (Recitals 52 and 52(a) of the compromise text of a Directive). The EMA and NCAs should actively support the design of comparative trials when providing pre-authorisation advice (Recital 52a of the compromise text of a Directive).
In addition, the EMA is enabled to consult various experts and authorities when needed in preparing scientific advice, including on clinical trials aspects:
- Member State clinical trial experts (Article 58.2 of the compromise text of a Regulation).
- Relevant NCAs and bodies for the exchange of information on general issues of scientific or technical nature (Article 162.1 of the compromise text of a Regulation). This process expressly covers guidelines on clinical trial design and evidence generation across the medicine lifecycle. It encompasses:
- HTA bodies and pricing and reimbursement authorities with the aim that such dialogue serves to align pre-authorisation regulatory guidance with the broader evidence needs of downstream decision-makers. (Recitals 38 and 39 of the compromise text of a Directive)
- Authorities or public bodies established under other relevant EU legal acts, including experts in clinical trials, medical devices, or substances of human origin, as required for the scientific advice in question. (Recital 65 of the compromise text of a Regulation). Recitals 45 and 45(a) also highlight the need for submission of raw clinical trial data and attention to trial composition, including gender-based equity.
The EMA's scientific advice function therefore forms part of a multi-authorities and experts consultation process that goes far beyond the simple one regulator- one sponsor relationship. The importance of early regulatory interaction and support mechanisms for medicines reinforce a model in which clinical development is guided through iterative dialogue with regulatory authorities. More information on scientific advice on EuroGCT Support from several competent authorities for the development of innovative medicines.
Regulatory sandboxes: The compromise text of a Regulation introduces a regulatory sandbox mechanism with direct implications for clinical research. Under Article 113.2, a sandbox establishes a controlled regulatory framework allowing targeted adaptations to certain requirements of the EU legislation to the extent strictly necessary to assess whether a marketing authorisation can be granted for the concerned medicine. With this process, the core principles of quality, safety and efficacy must be maintained, and the sandbox operates under the direct supervision of the competent authorities of the Member States concerned. The clinical trial dimension is addressed in Article 114.1 which provides that Member States shall take the sandbox plan into consideration when authorising a clinical trial application for products covered by a sandbox.
The sandbox under Recital 135 is a time-limited experimentation space designed to generate regulatory learning while preserving accountability. The supervisory powers of competent authorities and the liability of participants, including clinical trial sponsors, remain fully in force throughout the process. The sandbox outcomes are expressly envisaged as a source of input for future legislative evolution as the Commission is empowered to develop adapted regulatory frameworks on the basis of sandbox results. The sandbox thus operates as a structured pathway for evidence-based regulatory adaptation, enabling the testing of innovative medicines or regulatory approaches within a controlled environment under regulatory supervision.
- Clinical trials involving Genetically Modified 0rganisms (GMO)-based medicines: The Pharmaceutical Package also addresses a long-standing regulatory bottleneck affecting clinical trials with investigational medicines containing or consisting of GMOs. Recitals 145 to 152 of the compromise text of a Regulation acknowledge that the current framework, which requires sponsors to comply with divergent national procedures under Directives 2001/18/EC and 2009/41/EC, generates significant delays particularly for multi-centre trials. The Regulation therefore amends Regulation (EU) No 536/2014 to establish a centralised Environmental Risk Assessment (ERA) procedure, coordinated by a Reporting Member State and supported where relevant by the EMA's Environmental Risk Assessment working party. More information on Genetically Modified Organisms-Based Advanced Therapy Medicinal Products | EuroGCT.
Paediatric clinical trials: The compromise text of a Regulation also reinforces the framework governing clinical trials in the paediatric population. Recital 106 precises that medicines, before to be marketed within the EU, must be studied in children to ensure they are appropriately authorised for paediatric use and presented in suitable dosages and formulations. Recital 110 preserves a proportionate approach: the obligation to conduct paediatric studies may be waived where the medicine is likely to be ineffective or unsafe for children, where it offers no significant therapeutic benefit over existing options, or where the disease concerned occurs only in adult populations. The obligation is maintained where the product's mechanism of action is relevant to a different paediatric condition within the same therapeutic area.
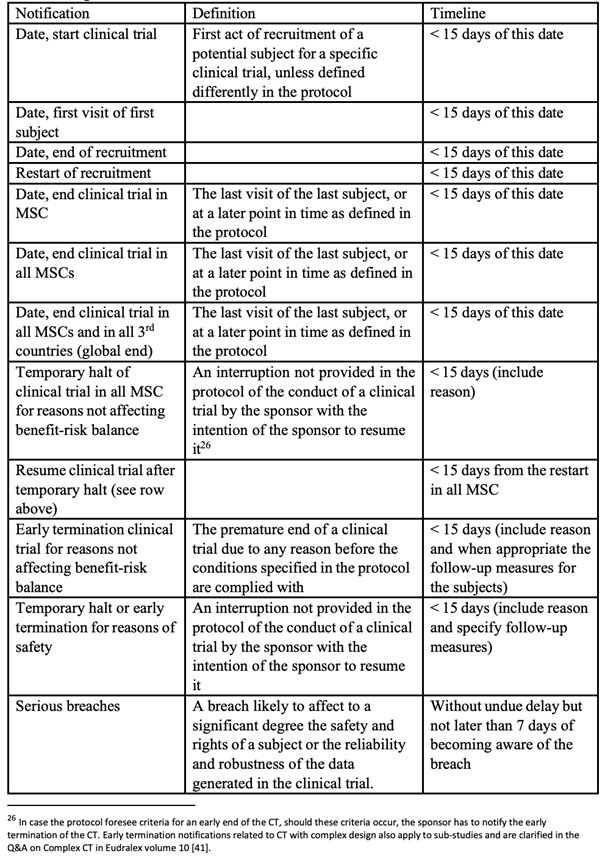
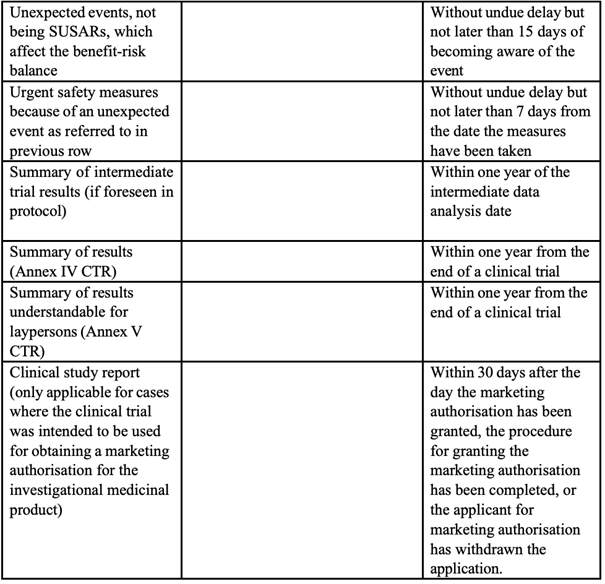
On the transparency side, Recitals 122 and 123 and Article 94 of the compromise text of a Regulation, introduce enhanced reporting obligations for paediatric clinical trials conducted in third countries. Where such trials are referenced in an agreed paediatric investigation plan, their key elements, i.e. trial protocol, investigational medicines, therapeutic indications and trial population, must be registered in the EU clinical trials database prior to the start of the trial, and a summary of results made publicly available within six months of completion, extendable to twelve months for justified scientific reasons. Article 177.4 of compromise text of a Regulation also amends Regulation (EU) No. 536/2014 directly, inserting into Article 37.4 a specific six-month deadline for the submission of results summaries to the EU database in all clinical trials involving medicines in the paediatric population. These provisions collectively strengthen the transparency framework of paediatric clinical development, reinforcing the obligation to generate and disclose robust data on medicines, including ATMPs, for children.
Clinical trials conducted in third countries: The compromise text of a Regulation also consolidates the conditions under which clinical trials conducted outside the EU may support a centralised marketing authorisation application. Recital 52 states that where clinical trials have been carried out in third countries on medicines intended for a centralised marketing authorisation, it must be verified, at the time of the marketing authorisation evaluation, that those trials were conducted in accordance with principles equivalent to those of Regulation (EU) No. 536/2014. This verification covers both the rights and safety of participants and the reliability and robustness of the data generated in the clinical trial. This requirement is stated in Article 6.1 which requires that marketing authorisation applications include an explicit declaration confirming that clinical trials carried out outside the Union meet the ethical requirements of Regulation (EU) No. 536/2014. Article 94(4) further empowers the Commission to adopt implementing acts to refine, on the basis of operational experience, the details to be submitted to the EU database concerning third-country trials.
These provisions ensure that clinical data generated outside the EU is subject to consistent ethical and transparency standards as a condition of regulatory acceptance within the EU.
Processing personal data: Under Article 169.1 of the compromise text of a Regulation, the EMA is empowered to process personal health data from clinical trials and other sources including real-world data for the purpose of improving the robustness of its scientific assessment or verifying claims of the applicant or marketing authorisation holder. This power also extends to broader regulatory science activities, provided that two cumulative conditions are met: the processing must be strictly necessary and proportionate to the objectives pursued, and where sensitive health data are involved, appropriate technical and organisational safeguards must be in place to protect data subjects' rights in line with EU law.
Overall, while the precise implementation details will depend on the final legislative text and subsequent regulatory guidance, the EU Pharmaceutical Package does not fundamentally alter the legal framework governing the conduct of clinical trials, which remains governed by Regulation (EU) No 536/2014. However, it provides for some changes. Through a combination of incentive mechanisms, enhanced scientific guidance and innovation-support tools such as regulatory sandboxes, the reform is likely to promote more robust, strategically designed and regulatorily aligned clinical trials across the EU. See also the impact of the pharmaceutical reform on the clinical trials of GMO-based ATMP, on EuroGCT Genetically Modified Organisms-Based Advanced Therapy Medicinal Products.
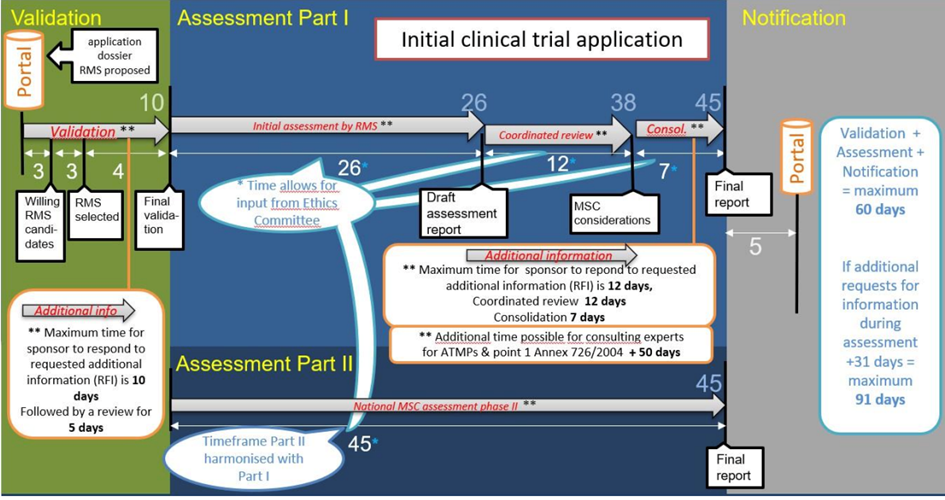
4. It currently exists specific rules concerning clinical trials of advanced therapy investigational medicinal products. They provide supplementary delays in order to consult experts for the assessment of the scientific part of clinical trials applications in view of ATMPs’ specificities:
a. For the assessment of initial clinical trials applications: The reporting Member State may extend the 45 days period to submit the final Part I of the assessment report to the sponsor and the other Member State by a further 50 days for clinical trials involving an advanced therapy investigational medicinal product for the purpose of consulting with experts. (Article 6§7 of Regulation (EU) No 536/2014).
The proposal of the European Biotech Act provides for a complete rewriting of the Article 6, with a significant reduction of timelines, including the deletion of this additional delay. (See below the Biotech Act section)
b. For the assessment of a substantial modification of an aspect covered by the scientific part of the assessment report: The reporting Member State may extend the 38 days period to submit, through the EU portal, the final assessment report including its conclusion to the sponsor and the other Member states concerned by a further 50 days for clinical trials involving an advanced therapy investigational medicinal product for the purpose of consulting with experts. (Article 18§5 of Regulation (EU) No 536/2014).
The proposal of the European Biotech Act provides for the deletion of this additional delay to reduce the clinical trial timelines. (See below the Biotech Act section)
5. Proposal for a new European Biotech Act:
The proposal for a new European Biotech Act is also proposing changes regarding clinical trials of ATMPs. The European Commission formally proposed the EU Biotech Act on 16 December 2025, following a public consultation held earlier that year. The legislation now enters the inter-institutional negotiation phase between the European Parliament and the Council, with final adoption anticipated in late 2026 or 2027. Consequently, entry into application is projected for the 2027–2028 period at the earliest.
The proposal introduces a targeted reform of the Clinical Trials Regulation (EU) No 536/2014, with significant implications for ATMP clinical trials. Its objective is to “simplify and future-proof the regulatory environment” and reduce time-to-market for innovative therapies, particularly in biotechnology and health. (Biotechnology - Public Health - European Commission).
The key amendments to Regulation (EU) No 536/2014, provided by Article 58 of the proposal for European Biotech Act, are the following:
a. Authorisation timelines for ATMP clinical trials: previously, certain additional assessment periods (+ 50 days) applied specifically to ATMPs, which lengthened the overall evaluation process. Under the Biotech Act, Article 6 of the Regulation (EU) No 536/2014 is amended to remove this additional period, thereby aligning the assessment of ATMP trials with standard timelines applicable to other clinical trials. This change is expected to reduce procedural delays for multinational ATMP studies and to accelerate the pace of clinical development.
b. Reduction of overall authorisation timelines by amending Articles 5 to 8 of the Regulation (EU) No 536/2014. These changes restructure the coordinated assessment procedure, shorten statutory decision deadlines, and limit delays associated with the review of substantial modifications. For instance, the maximum assessment period for multinational trials is reduced from 106 days to 75 days, while deadlines for reviewing protocol amendments are shortened from 96 to 47 days or from 64 to 33 days depending on the nature of the modification. These measures are designed to facilitate more efficient trial initiation and continuation, which is particularly relevant for the complex and iterative development of ATMPs.
c. The strengthening of the role of the reporting Member State by amending Articles 5 and 7 of the Regulation (EU) No 536/2014. By clarifying and enhancing the reporting Member State’s coordination responsibilities, the amendments aim to harmonise scientific and ethical evaluations across Member States. This is especially important for ATMP trials, where divergent national interpretations could otherwise create bottlenecks in multinational studies.
d. Facilitation of the use of previously submitted clinical trial data through an amendment to Article 11 of the Regulation (EU) No 536/2014. This provision enables sponsors to reference existing data for investigational products in related clinical trials, thereby reducing duplicative submissions and allowing for more efficient iterative development. This is particularly relevant for ATMPs, where repeated preclinical and early-phase clinical data may serve as a basis for successive trials in the development pathway.
e. Establishment of regulatory sandboxes to provide experimental frameworks in which ATMPs can be developed under controlled regulatory flexibility. They aim to support innovative clinical trial protocols, including combination therapies and novel manufacturing processes, while safeguarding patient safety and data integrity.
f. The European Biotech Act states also that “Communication between sponsors and Member States will be improved during assessments. A single, core dossier for investigational products will simplify clinical trials using the same investigational medicine and help the conduct of registration trials and the preparation of marketing authorisation applications in Europe. Simplifications for low-intervention clinical trials will be further supported by introducing a new category of ‘minimal-intervention’ clinical trials. Mandatory EU harmonised templates will enable harmonisation. A single assessment process will be defined for combined studies involving the investigation of a medicine together with a medical device or an in-vitro diagnostic. The legal basis for processing personal data in clinical trials in accordance with Regulation (EU) 2016/679 requirements will be harmonised. Accelerated and simplified procedures will enable multinational clinical trials to be carried out on in relation to public health emergencies”. (Amendments to Regulation (EU) No 536/2014 (Clinical Trials Regulation)
For an overview of the key amendments to Regulation (EU) No 536/2014, provided by Article 58 of the proposal for European Biotech Act, please see below an extract from a EuroGCT poster presented at ISCT 2026: A. MAHALATCHIMY, V. ROBY, C. CHABANNON, The European Commission proposal for a new regulation on biotechnology: why does it matter for ATMP? , ISCT 2026, 6-9 May 2026, Dublin, Ireland.
See also the impact of the European Biotech Act on the clinical trials of GMO-based ATMP, on EuroGCT Genetically Modified Organisms-Based Advanced Therapy Medicinal Products.
See also the European Commission’s Factsheet - How Clinical Trials will change under the Biotech Proposal.
In conclusion, the European Biotech Act represents a transformative set of reforms for ATMPs clinical trials in the EU in particular by removing ATMP-specific procedural extensions and reducing overall timelines. The Act creates a more predictable and innovation-friendly environment to accelerate the development of advanced therapies while maintaining high standards of patient safety, scientific requirements, and regulatory transparency.

See also the impact of the European Biotech Act on the clinical trials of GMO-based ATMP, on EuroGCT Genetically Modified Organisms-Based Advanced Therapy Medicinal Products.
See also the European Commission’s Factsheet - How Clinical Trials will change under the Biotech Proposal.
In conclusion, the European Biotech Act represents a transformative set of reforms for ATMPs clinical trials in the EU in particular by removing ATMP-specific procedural extensions and reducing overall timelines. The Act creates a more predictable and innovation-friendly environment to accelerate the development of advanced therapies while maintaining high standards of patient safety, scientific requirements, and regulatory transparency.