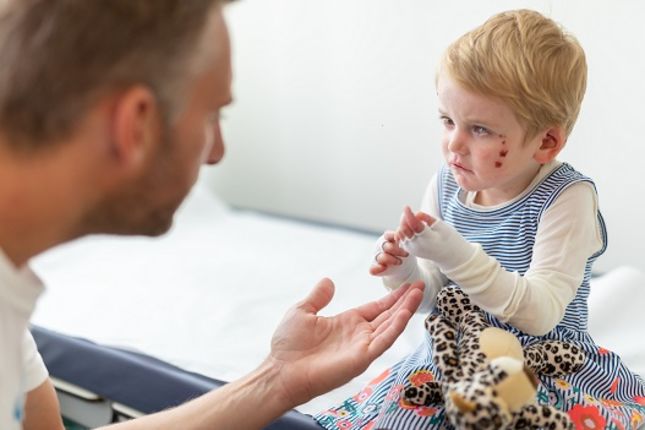
La Epidermólisis Bullosa (EB) es una enfermedad genética que afecta a la cohesión de las células y tejidos. Hay varias formas de EB, pero un síntoma común a todas ellas es una piel muy frágil que fácilmente se desgarra o desarrolla llagas y ampollas incluso debido a una mínima fricción, estrés o daño. Actualmente no existe cura para la EB, pero ya se está utilizando la terapia génica para tratar a personas con un tipo específico de EB, y hay investigaciones y ensayos clínicos prometedores que están explorando nuevas formas en que la terapia génica y celular pueden ayudar.
¿Qué sabemos?
Todas las formas de EB están causadas por mutaciones en genes que afectan a la forma en que las células y los tejidos se conectan entre sí. La EB simple, la EB juncional, la EB distrófica y la EB de Kindler son los cuatro tipos principales de EB.
La EB es relativamente rara, con una incidencia estimada de 1 de cada 50 000 nacidos vivos.
La EB afecta visiblemente a la piel, pero puede causar complicaciones en otros órganos como, por ejemplo, dar lugar a la aparición de llagas y ampollas en la garganta, las vías aéreas superiores y el tracto urinario. Además, puede resultar en una enfermedad sistémica progresiva con consecuencias incapacitantes y puede resultar en cánceres de piel agresivos.
Actualmente no existe cura para la EB, pero recientemente se ha aprobado una terapia génica tópica para ayudar a tratar las heridas causadas por un tipo específico de EB.
¿Qué se está investigando?
Hay muchos dispositivos, fármacos y tratamientos biológicos nuevos que se están estudiando en ensayos clínicos. Muchos de estos estudios tienen como objetivo mejorar el tratamiento de la EB. Algunos estudios tienen como objetivo «corregir» las mutaciones genéticas subyacentes que causan la EB.
El desarrollo de nuevas terapias génicas podría permitir restaurar los genes sanos de las células de la piel. El trabajo con células madre epidérmicas (de la piel), la optimización de las técnicas de cultivo de piel en el laboratorio y la mejora de los métodos de injerto de tejidos son también componentes clave para el desarrollo de terapias génicas eficaces para la EB.
¿Cuáles son los retos?
El tratamiento de un trastorno genético es muy difícil porque la causa se encuentra en el ADN de la persona. Los tratamientos para compensar las mutaciones genéticas deben reparar el gen mutado o añadir una nueva copia funcional del gen a las células. No es una tarea sencilla. Modificar el ADN celular o añadir nuevos genes a las células conlleva riesgos potenciales, como la posibilidad de desarrollar cáncer. Los trasplantes de células y piel pueden conllevar riesgos como hemorragias, infecciones y rechazo del injerto.
La investigación clínica minuciosa tiene como objetivo garantizar que los nuevos tratamientos para la EB sean seguros y eficaces.
Otro reto pendiente es hacer que los tratamientos complejos, como las terapias génicas, sean asequibles y accesibles para todo el mundo.
Acerca de la epidermólisis bullosa
La epidermólisis bullosa (EB) es un grupo de trastornos genéticos raros del tejido conectivo que provocan una piel muy frágil y con ampollas. Las ampollas pueden estar causadas por la fricción de la piel, lesiones cutáneas leves y actividades cotidianas, como frotarse o rascarse. Estas ampollas pueden convertirse en llagas dolorosas y heridas abiertas. En las formas graves de EB, las ampollas y las llagas también se desarrollan en otros tejidos, como la boca, la garganta, el estómago, las vías respiratorias superiores y las vías urinarias.
Existe una amplia gama de gravedad de la EB. Los tipos leves de EB pueden provocar ampollas leves a lo largo de la vida, y otros solo presentan síntomas graves de EB durante la infancia, que pueden mejorar con la edad. Algunas formas de EB pueden poner en peligro la vida debido a la pérdida de grandes cantidades de piel. Otras pueden causar cicatrices extensas, fusión de dedos de manos y pies, así como otras complicaciones que limitan la vida. Algunas formas de la enfermedad pueden conducir al desarrollo de cáncer de piel, lo que reduce la esperanza de vida.
La EB es poco frecuente, lo que dificulta la estimación del número total de personas que la padecen. DEBRA, una organización benéfica dedicada a la EB, estima que alrededor de 500 000 personas en todo el mundo padecen algún tipo de EB.
Todos los tipos hereditarios de EB están causados por mutaciones en el ADN de la persona afectada. Estas mutaciones alteran uno de los varios genes importantes para la producción de proteínas que conectan las células y los tejidos. Por eso, la EB también se conoce como trastorno del «tejido conectivo». Actualmente hay 16 genes en nuestro ADN que se han relacionado con la EB hereditaria clásica.
¿Cómo se hereda la EB? Una mutación genética relacionada con la EB suele transmitirse de padres a hijos. Nuestro ADN contiene dos copias de cada gen (excepto los genes de los cromosomas X e Y). Varias formas de EB solo requieren heredar una copia de una mutación genética relacionada con la EB. Esto se conoce como forma «dominante» de EB. El progenitor que transmite una forma dominante de mutación genética relacionada con la EB también padece EB y sus síntomas. Otras formas de EB requieren heredar una mutación genética relacionada con la EB de ambos progenitores. Estas se denominan formas «recesivas» de EB. Las formas recesivas de EB suelen ser una sorpresa para la familia, ya que ninguno de los progenitores presenta los síntomas de la EB y es posible que no sepan que son portadores de la mutación.
Acerca de la piel
Nuestra piel es un órgano extenso y complejo. Actúa como un escudo resistente, flexible e impermeable que mantiene el agua dentro y las bacterias fuera. Nos protege del viento y las inclemencias del tiempo y nos ayuda a regular la temperatura corporal. Es donde se sintetiza la vitamina D. Además de todo esto, nuestra piel tiene una gran cantidad de células especializadas que nos permiten sentir la temperatura, las texturas, la presión y el dolor.
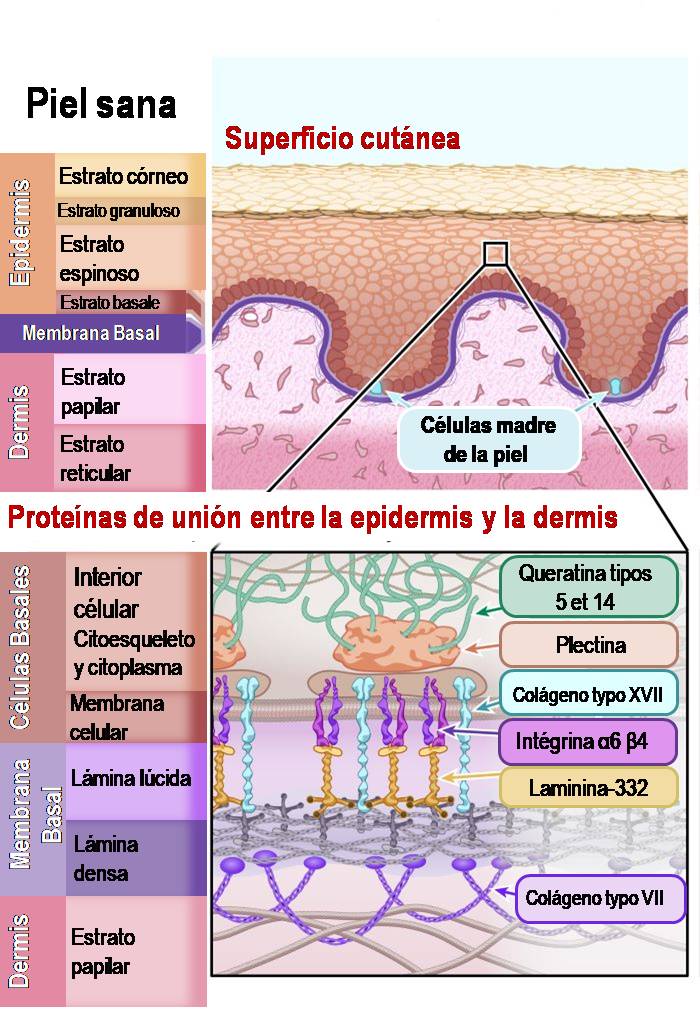
Las dos capas principales de la piel son la epidermis (la capa más externa) y la dermis. Los investigadores subdividen estas capas en capas específicas (o estratos) de células. Puedes ver estas subcapas en el siguiente diagrama.
La epidermis está compuesta principalmente por células llamadas queratinocitos. Se llaman queratinocitos por la proteína resistente que producen, llamada queratina, que es la misma proteína que forma nuestro cabello y nuestras uñas. Los queratinocitos son producidos por células madre de la piel que se encuentran en la subcapa del estrato basal de la epidermis y en los folículos pilosos de la dermis. Las células madre de la piel producen constantemente nuevos queratinocitos para reemplazar las células cutáneas que se caen o se desprenden continuamente. Esto hace que las células madre de la piel sean muy importantes para mantener la salud de nuestra piel.
La dermis es la capa de la piel que contiene los vasos sanguíneos, las células sensoriales del tacto, la temperatura y el dolor, las glándulas sudoríparas, los folículos pilosos y mucho más. Hay muchos tipos diferentes de células en la dermis, pero la mayor parte de la dermis está formada en realidad por una malla flexible de fibras conectivas y proteínas. Esta malla de proteínas sostiene la estructura de la piel y mantiene todo en su sitio. Las proteínas que intervienen en esta malla son principalmente el colágeno y la elastina.
El punto de conexión entre la epidermis y la dermis también es importante, especialmente cuando se habla de EB. La «membrana basal» es una fina capa de tejido conectivo importante para unir la epidermis y la dermis. La membrana basal está compuesta principalmente por dos tipos de proteínas, la laminina y el colágeno.
Hay cuatro tipos principales de EB: EB simple (EBS), EB juntural (EBJ), EB distrófica (EBD) y EB de Kindler (EBK). Estas categorías se distinguen comúnmente por la capa de la piel donde se forman las ampollas. Aunque todas las formas de EB presentan ampollas como síntoma, no están causadas por el mismo problema. El gen afectado por una mutación genética relacionada con la EB determina dónde, por qué y en qué medida se producen las ampollas y el desprendimiento de la piel. También determina el tipo de EB que padece una persona.
Actualmente se han identificado 16 genes específicos en nuestro ADN que están relacionados con la EB clásica. Las mutaciones en estos genes impiden que las células produzcan proteínas funcionales importantes que mantienen unidas la epidermis y la dermis. Conocer la función que desempeñan normalmente las proteínas en la unión de la piel es fundamental para comprender los diferentes tipos de EB. También revela por qué algunos tipos de EB son más graves que otros y por qué un tratamiento para un tipo de EB puede no funcionar para todos.
A continuación se presentan resúmenes que destacan los genes, sus proteínas y algunas características específicas de dónde y por qué se produce el desprendimiento en tres de las principales formas de EB.
EB Simple:
La EB simple es la forma más común de EB y representa alrededor del 70 % de todos los casos de EB. La gravedad de los síntomas de la EBS puede variar, desde fragilidad cutánea y ampollas leves en las manos y los pies, hasta casos en los que se producen ampollas en todo el cuerpo, pasando por subtipos más graves. La EBS se hereda predominantemente de los padres como un rasgo dominante, lo que significa que solo uno de los padres necesita transmitir la mutación genética relacionada con la EB. Ese progenitor tiene EBS y es probable que tenga (o haya tenido) los síntomas asociados.
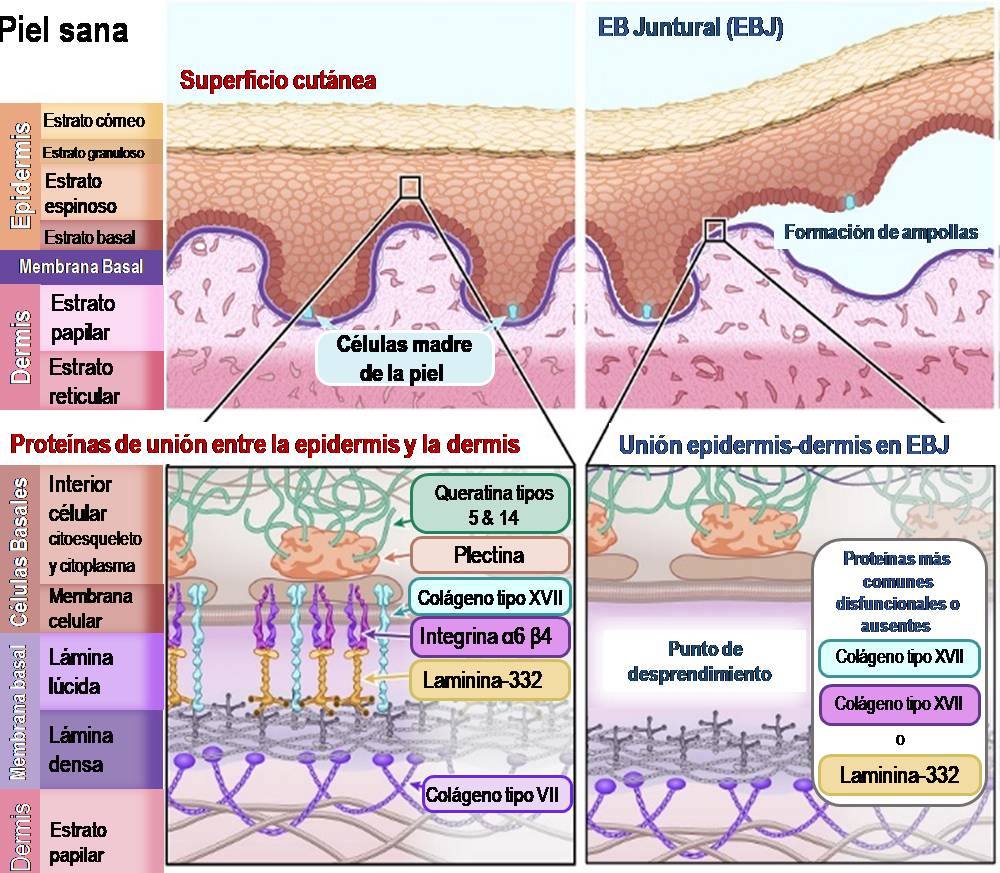
En la EBS, la separación de la epidermis y la dermis se produce dentro de las células basales de la epidermis (estrato basal), justo por encima de la membrana basal. El problema es que las proteínas que fijan las células basales a la membrana basal no se adhieren correctamente al soporte estructural dentro de la célula (el citoesqueleto). Esto provoca que las células basales se desgarren y se rompan, dejando la parte inferior de estas células adherida a la membrana basal.
Las principales proteínas afectadas por las mutaciones genéticas en la EBS son:
Queratina tipo 5 – [Nombre del gen: KRT5]
Queratina tipo 14 – [Nombre del gen: KRT14]
Plectina – [Nombre del gen: PLEC1]
Ilustración 2: Esquema de piel sana en comparación con piel afectada por Epidermólisis Bullosa Simplex.
EB juncional :
La EB juncional (EBJ) constituye aproximadamente el 10 % de todos los casos de EB. La EBJ se hereda de los padres como un rasgo recesivo, lo que significa que una persona debe haber heredado una mutación genética relacionada con la EB de ambos padres. Es probable que esos padres no presentaran ningún síntoma de EB.
Algunas personas afectadas por la EBJ padecerán «EBJ grave» (antes denominada EBJ generalizada grave o EBJ de Herlitz). Las ampollas se extienden por todo el cuerpo, incluyendo la boca, la nariz y la garganta. Estas ampollas impiden que los recién nacidos coman y respiren correctamente, lo que provoca desnutrición e insuficiencia pulmonar. Trágicamente, este subtipo extremo de EBJ es mortal y muy pocos niños con EBJ grave viven más de dos años.
El otro subtipo principal es la «EBJ intermedia» (antes llamada EBJ generalizada intermedia o EBJ no Herlitz). Los síntomas de este grupo son menos graves, pero siguen siendo intensos y, lamentablemente, las muertes siguen siendo frecuentes. Las personas con EBJ intermedia pueden llegar a la edad adulta, pero a menudo presentan grandes cantidades de cicatrices, problemas en las uñas y los dientes, así como otras complicaciones que limitan su esperanza de vida.
En la EBJ, la separación de la epidermis y la dermis se produce en la membrana basal, concretamente en la subcapa «lámina lucida». Esta subcapa está formada por proteínas que anclan las células basales de la epidermis a la dermis. Cuando las proteínas de esta capa faltan o no funcionan, este punto de unión de la membrana basal no se mantiene unido.
Las principales proteínas afectadas por las mutaciones genéticas en la EBJ son:
Colágeno tipo XVII (también conocido como BPAG2) – [Nombre del gen:COL17A1]
Integrina α6β4 (subunidades alfa 6, beta 4) – [Nombres de los genes: ITGA6 e ITGB4]
Laminina-332 (también conocida como laminina-5) – [Nombres de los genes: LAMA3, LAMB3 y LAMC2]
Ilustración 3: Esquema de piel sana en comparación con piel afectada por Epidermólisis Bullosa Juntural.
EB distrófica:
La EB distrófica (EBD) representa alrededor del 20 % de todos los casos de EB. La EBD puede heredarse como un rasgo dominante o recesivo, dependiendo de la mutación genética específica.
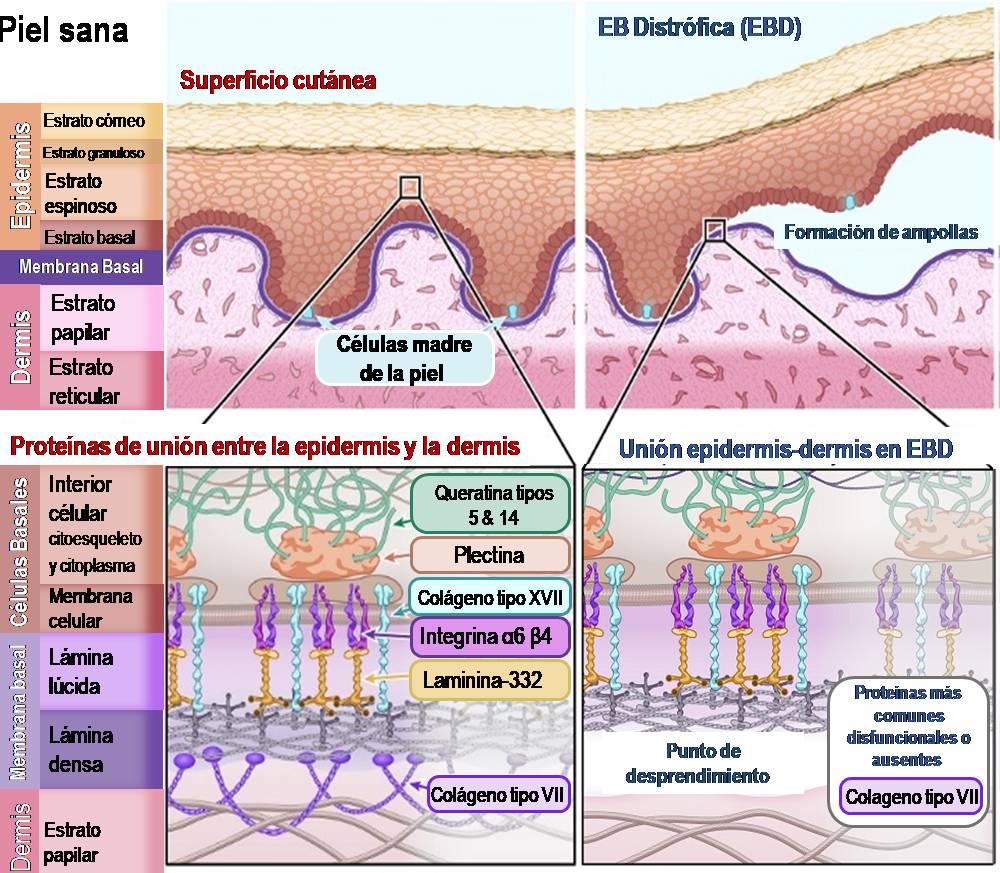
n la EBD, la separación de la epidermis y la dermis se produce bajo la membrana basal. La proteína principal que une la membrana basal a la dermis es el colágeno VII. Esta proteína forma fibrillas de anclaje que se entrelazan con las proteínas estructurales de la dermis. Sin colágeno VII, la membrana basal se desprende fácilmente de la dermis.
La proteína principal afectada por una mutación genética en la DEB es:
Colágeno tipo VII – [Nombre del gen: COL7A1]
Ilustración 4: Esquema de piel sana en comparación con piel afectada por Epidermólisis Bullosa Distrófica.
EB de Kindler:
La EB de Kindler (KEB) es un subtipo raro de EB hereditaria. La KEB es una forma recesiva de EB, lo que significa que una persona hereda la mutación genética de ambos padres. Los síntomas pueden variar de leves a graves e incluyen fragilidad cutánea, sensibilidad a la luz, decoloración y engrosamiento de la piel. La enfermedad de las encías, las ampollas en la boca y la inflamación intestinal pueden afectar a la alimentación, y las personas con KEB tienen un mayor riesgo de desarrollar cáncer de piel.
La KEB puede afectar a diferentes y múltiples capas de la piel. Cuando la proteína afectada falta o no funciona, se altera la capacidad de los queratinocitos (células de la piel) para crecer y dividirse normalmente, y para unir la epidermis a la dermis.
La principal proteína afectada por una mutación genética en la KEB es:
Kindlin-1 – [Nombre del gen: FERMT1]
Nuestros socios de la Red de Investigación sobre la EB disponen de información sobre la EB y sus formas.
Los tratamientos se centran principalmente en prevenir o aliviar los síntomas de la EB. Las medidas preventivas pueden incluir el uso de vendajes y almohadillas para minimizar la fricción, los golpes y los traumatismos leves en la piel. Los tratamientos también pueden tener como objetivo prevenir infecciones y ayudar a que las ampollas y las llagas se curen.
En los casos más graves de EB, se pueden utilizar medicamentos para prevenir infecciones de heridas abiertas y llagas. También se puede recurrir a la cirugía para evitar que los dedos de las manos y los pies se fusionen o que la garganta y el esófago (tubo por donde pasa la comida) se estrechen demasiado.
En casos extremos de EB, los médicos pueden cubrir las heridas abiertas grandes mediante injertos de piel. Sin embargo, los injertos de piel de donantes son muy poco frecuentes, ya que el sistema inmunitario casi siempre rechaza los injertos de piel de donantes, incluso si el paciente y el donante son muy compatibles. Por ello, los médicos suelen utilizar sustitutos cutáneos temporales para cubrir las heridas abiertas grandes. También se están generalizando los nuevos «sustitutos biológicos de la piel».
Estos sustitutos cutáneos avanzados tienen como objetivo mejorar la cicatrización de las heridas mediante el uso de mallas sintéticas en las que las células pueden crecer, proteínas presentes en la piel y, en ocasiones, incluso queratinocitos vivos.
Terapia génica para la EB distrófica
Actualmente existe una terapia génica aprobada en la UE que se dirige a la causa genética subyacente de la EB distrófica. Vyjuvek es una terapia génica tópica, lo que significa que se aplica directamente sobre la piel en forma de gel. Se utiliza para ayudar a curar las heridas y lesiones que se producen a causa de la EB.
Vyjuvek actúa administrando dos copias sanas del gen COL7A1 a los queratinocitos y fibroblastos de la piel, lo que les permite producir colágeno y favorecer la cicatrización de las heridas. Vyjuvek administra el gen COL7A1 a las células utilizando una forma inactivada del virus del herpes simple (VHS-1) como vector. El VHS-1 es el mismo virus que causa el herpes labial. Sin embargo, estas partículas víricas han sido modificadas para impedir su replicación, lo que las hace seguras y evita que causen infecciones.
El HSV-1 no puede atravesar la barrera cutánea, por lo que este tratamiento solo puede aplicarse en heridas abiertas o lesiones. Esto significa que solo puede ayudar a curar las heridas, pero no puede impedir que se desarrollen. El tratamiento no es permanente, por lo que es necesario aplicarlo de forma continuada para mantener su eficacia clínica.
Investigación actual
Mejorar la calidad de vida
Hay muchos investigadores, empresas farmacéuticas y empresas de suministros médicos que trabajan para desarrollar mejores productos que mejoren el tratamiento de los síntomas de la EB.
Los productos que se están estudiando van desde vendajes y apósitos mejorados hasta pomadas y lociones que ayudan a la piel a curarse.
Crear piel «nueva»
La forma ideal de corregir un trastorno genético es corregir o sustituir un gen mutado en el ADN de una persona. Los investigadores están creando terapias génicas que hacen precisamente eso. Los nuevos descubrimientos y tecnologías, como la edición genética CRISPR-Cas9, están acelerando enormemente la forma en que se puede editar el ADN y los genes en las células. Sin embargo, cambiar el ADN de las células de una persona no es una tarea sencilla. El proceso también puede conllevar riesgos graves. Si la adición o edición de un gen sale mal, puede provocar cáncer, añadir nuevas complicaciones o empeorar el estado de la persona. Los ensayos clínicos son importantes para identificar los riesgos de los nuevos tratamientos y demostrar que realmente funcionan.
En muchos casos, las células madre van de la mano de las terapias génicas. Para las terapias génicas de la EB, los investigadores quieren editar el ADN de las células madre de la piel, en lugar de otros tipos de células cutáneas. Esto se debe a que añadir (o editar) un gen en las células madre de la piel hará que ese gen se transmita a todas las demás células que producen las células madre. Esto es importante porque casi todos los queratinocitos de nuestra piel se renuevan cada 4-5 semanas. Además, las células madre de la piel se renuevan continuamente, por lo que cualquier modificación del ADN introducida en una célula madre podría ser «permanente» (o durar al menos mientras sobrevivan las células madre).
Varios ensayos clínicos están probando métodos para editar el ADN de las células madre de la piel de pacientes con EB con el fin de cultivar «nueva» piel sana para injertos. Aunque los métodos y los detalles de estos diferentes ensayos clínicos varían, la idea general y el proceso son básicamente los mismos. Se toman muestras de piel de una persona afectada por la EB y se cultivan en un laboratorio; se espera que estas muestras contengan células madre de la piel. A continuación, se utilizan métodos de edición del ADN para añadir o editar genes en las células y restaurar la producción normal de proteínas. Las células madre de la piel con las nuevas ediciones del ADN se utilizan en métodos avanzados de cultivo celular para hacer crecer capas de epidermis en el laboratorio. A continuación, estas capas cultivadas en el laboratorio pueden injertarse en el paciente.
Algunos de estos enfoques implican el uso de células madre pluripotentes inducidas (iPSC), un tipo de célula madre que puede generarse a partir de células de nuestro cuerpo. Estas células pueden propagarse (autorrenovarse) indefinidamente y dar lugar a cualquier otro tipo de célula de la piel. Podrían representar una fuente única de células para sustituir el tejido dañado.
Este tipo de tratamiento requiere mucho tiempo y trabajo para identificar la mutación genética de cada persona afectada por la EB, desarrollar un enfoque específico de edición del ADN, cultivar células durante semanas y, finalmente, realizar la cirugía clínica para llevar a cabo los injertos de piel. También requiere una investigación muy especializada e instalaciones para el cultivo de células. A pesar de estos obstáculos, se han registrado importantes avances que demuestran que este proceso puede funcionar. En la siguiente sección se analiza uno de estos casos de éxito.
Foco en la investigación europea: una historia de éxito
En 2017, un grupo de investigadores y médicos dirigido por el profesor Michele De Luca informó del éxito de la terapia génica y los injertos de piel cultivada en laboratorio para salvar a un niño que había perdido más del 80 % de su piel. El niño padecía EB causada por mutaciones en el gen de la laminina (LAMB3) que había heredado de ambos padres. Todos los tratamientos, incluidos varios enfoques extremos, habían fracasado. Los médicos pensaban que era poco probable que el niño sobreviviera. Las autoridades gubernamentales concedieron permiso para el «uso compasivo» de un tratamiento preliminar de terapia génica que solo se había probado anteriormente en dos estudios de casos que analizaban a un solo paciente, respectivamente. Los padres del niño también aceptaron probar el procedimiento, incluso después de que se les informara de que el niño podría no sobrevivir al procedimiento en sí.
El niño sobrevivió y ahora vive con piel creada a partir de células editadas genéticamente en más del 80 % de su cuerpo. Los investigadores examinaron esta piel y afirman que se parece notablemente a la piel normal en muchos aspectos.
El éxito de este tratamiento es muy emocionante; algunos incluso podrían decir que el resultado fue milagroso, ya que superó las expectativas.
Lamentablemente, los tratamientos de terapia génica como estos aún no están aprobados clínicamente ni están ampliamente disponibles. La medicina personalizada como esta es cara, requiere instalaciones especiales y el trabajo de muchas personas. Esperemos que tratamientos como este se aprueben clínicamente, sean asequibles y estén ampliamente disponibles para muchas formas de EB.
El futuro es prometedor. El éxito del tratamiento de este niño, así como los resultados positivos de otros estudios, han dado lugar a varios estudios clínicos nuevos. M. De Luca, del Centro de Medicina Regenerativa «Stefano Ferrari» de Módena (Italia), junto con JW Bauer, del Hospital Universitario de Dermatología de Salzburgo (Austria), están dirigiendo varios estudios clínicos en los que se utilizan procedimientos similares, pero más avanzados, para sustituir las mutaciones de los genes COL7A1 y COL17A1 (Estudio 1, Estudio 2). Investigadores de otras instituciones también están desarrollando sus propias terapias génicas, como el ensayo clínico dirigido por Jean Yuh Tang, de la Facultad de Medicina de la Universidad de Stanford, que también se centra en las mutaciones subyacentes de la DEB en el gen COL7A1.
Otros enfoques terapéuticos
La creación de piel editada con ADN plantea grandes retos: el proceso es complejo, caro y arriesgado. Además, pueden pasar años antes de que este enfoque se convierta en un tratamiento realista.
Estos retos han llevado a los investigadores a pensar en otros enfoques para tratar la EB que puedan ser más rápidos de implementar, clínicamente aprobados y económicamente accesibles para las personas afectadas por la EB. Estos enfoques incluyen células madre, fármacos, extractos de plantas y muchos otros tratamientos. Los dos siguientes son solo algunos ejemplos.
Se están desarrollando cremas y lociones tópicas terapéuticas para contrarrestar los problemas de mutaciones específicas de la EB. Un tratamiento tópico, Vyjuvek, ya ha sido aprobado para su uso en la UE. Un estudio clínico de especial interés está utilizando una crema que intenta reparar el gen COL7A1, lo que ayudaría a las personas con EB distrófica. La crema contiene componentes que promueven que las células de la piel omitan la mutación del gen COL7A1 al producir la proteína colágeno VII.
Los trasplantes de médula ósea se utilizan para tratar la leucemia y otros trastornos de la sangre y del sistema inmunitario. Sin embargo, algunos grupos de investigación están explorando el tratamiento de la EB con trasplantes alogénicos de médula ósea, en los que las células madre sanguíneas proceden de donantes sanos. La ventaja de este enfoque es que los trasplantes de médula ósea se han utilizado con éxito durante muchos años y la técnica ha mejorado mucho a lo largo del tiempo. Los resultados preliminares sugieren que las células madre sanas de la médula ósea (o las células producidas por estas células madre) pueden producir proteínas del tejido conectivo que faltan en la piel, como el colágeno VII en personas con DEB. Sin embargo, este enfoque también tiene sus limitaciones, entre ellas la necesidad de un donante inmunocompatible, y actualmente los riesgos asociados son elevados.
En clinicaltrials.gov se puede encontrar una lista más completa de ensayos clínicos para la EB. (Tenga en cuenta que este sitio web público solo incluye una lista de ensayos clínicos. No comprueba si los ensayos clínicos incluidos son seguros, científicamente sólidos o realizados por instituciones de prestigio. Lea los avisos legales, consulte a profesionales sanitarios de confianza e infórmese sobre los riesgos y los posibles beneficios).
Find Out More
Recursos de DEBRA, una red de defensa y apoyo a los pacientes con EB:
EB Research Network: una alianza de organizaciones benéficas que apoyan la investigación sobre la EB, encabezada por DEBRA Austria