. Determination of the product nature
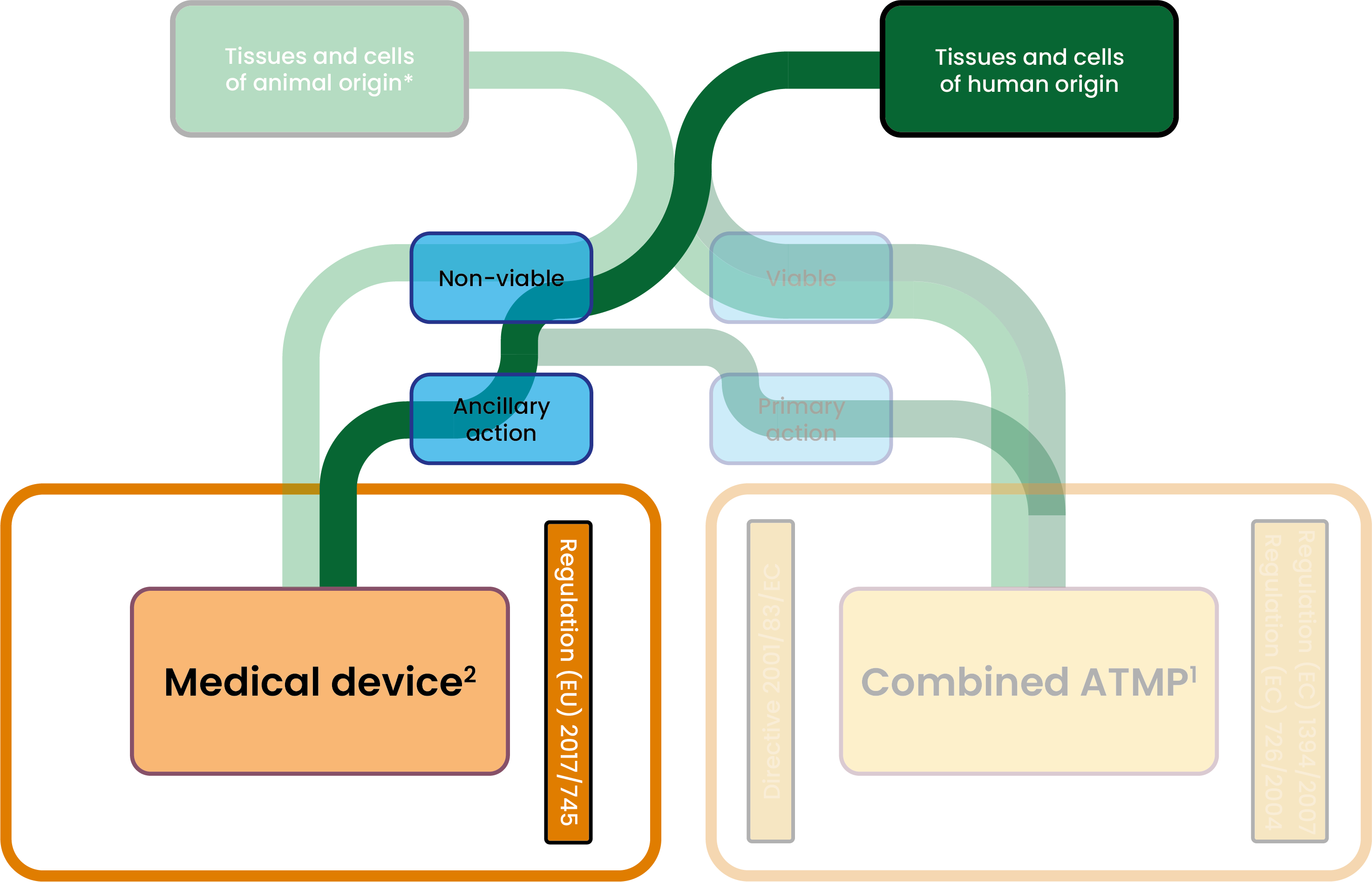
1 Article 2(1)(d) of the Regulation (EC) No 1394/2007 on ATMPs
2 Article 1(10) §1 of the Regulation on medical devices
2. Determination of the medical device class
The regulation on medical devices is structured around a classification of medical devices based on the risks, from devices of lesser risk, class I, to devices of higher risk, class III. The higher the class, and thus the risk, the stricter are the requirements to place them in the market.
A device’s classification is hence crucial as it will determine the applicable requirements.
3. Identification and selection of the conformity assessment procedure
In order to put a CE marking on a medical device, as required to place it on the market (Article 20 of the Regulation on medical devices), a medical device must undergo one of the conformity assessment procedures (Article 52 of the Regulation on medical devices). This assessment is performed by a Notified body for all medical devices except for class I devices, and aims at ensuring the manufacturer complies with regulatory requirements.
As already mentioned, the class of a medical device is decisive in determining the requirements that will apply to it. The same therefore applies to the conformity assessment procedure.
For class III medical devices (all medical devices manufactured using non-viable substances of human origin):
The conformity assessment will require the involvement of a notified body. To this end, the manufacturer is free to choose and may submit its application for assessment to any notified body within the EU, designated by a national authority responsible for notified bodies (see the List of notified bodies or Actors and Network database).
There is only one condition governing the manufacturer’s choice of notified body: “provided that the chosen notified body is designated for conformity assessment activities related to the types of devices concerned”. (Article 53(1) of the Regulation on medical devices)
Before submitting an application for conformity assessment to a notified body, the manufacturer of a Class III medical device shall assign a unique device identifier (UDI-DI) to the device and register it. (Article 29(3) of the Regulation on medical devices)
Commission Implementing Regulation (EU) 2026/977 provides certain uniform quality management and procedural requirements for the conformity assessment activities carried out by a notified body (mainly quotations, timelines and re-certification).
The manufacturer then has two options regarding assessment by a Notified Body:
- Conformity assessment based on a quality management system and assessment of the technical documentation (Annex IX of the Regulation on medical devices)
- Conformity assessment based on type examination (Annex X) and product conformity verification (Annex XI)
Conformity assessment based on a quality management system and assessment of the technical documentation (Annex IX of the Regulation on medical devices)
This assessment comprises both an evaluation of the quality management system and an evaluation of the technical documentation.
Assessment of the quality management system by the chosen notified body (NB):
Following the manufacturer’s submission of an application for assessment of the quality management system, the NB assesses the manufacturer’s quality objectives, the measures taken, the organisation and the personnel qualification, the control and surveillance systems from the conception of the devices to their production. As part of this assessment, the NB conducts an on-site audit, at the manufacturer’s premises and those of its suppliers and/or subcontractors, where applicable, in order to control the Quality Management System (QMS) and its effective application.
Once this assessment has been carried out, and if the QMS is in compliance with the regulation, the NB issues an EU quality management system certificate, first step to the CE marking of conformity. (Annex IX-2 of the Regulation on medical devices)
Assessment of the technical documentation by the chosen notified body (NB):
Following the manufacturer’s submission of an application for assessment of the technical document, the NB also conducts a thorough review of the technical documentation described in the Obligations applicable to medical device manufacturers. This control consists in the assessment of the evidence provided by the manufacturer, of the risk-benefit ratio, and where applicable, of the equivalence with an already existing device, all of it taking into account the innovative character of the medical device, the risk management plan, the target population, the post-market surveillance plan and the post-market clinical follow-up (see Obligations applicable to medical device manufacturers). (Annex IX-4.1 to 4.8 of the Regulation on medical devices)
Once this assessment has been carried out, and if the device complies with the regulation, the NB issues an EU technical documentation assessment certificate. (Annex IX-4.9 of the Regulation on medical devices)
It should be noted that there are additional special procedures for certain medical devices:
More specifically, for medical devices manufactured using non-viable tissues or cells of human origin (i.e. Class III): Before taking its decision, the NB must consult one of the competent authorities for human tissues and cells within the meaning of the Directive 2004/23/EC / SoHO authority in accordance with Regulation (EU) 2024/1938. On the basis of the preliminary assessment of the technical documentation containing the relevant information on these tissues and cells, the competent authority to which the request is made issues an opinion on the various aspects relating to the substances of human origin.
If the competent authority issues a negative opinion, the NB cannot issue a certificate of conformity. In the event of a favourable opinion, the NB retains the possibility of issuing such a certificate or refusing to do so, provided, however, that it takes into account the views expressed in that opinion and incorporates it into its documentation relating to the medical device. The NB is also required to communicate its final decision to the competent authority concerned. (Annex IX-5.3.1 of the Regulation on medical devices)
The Regulation on medical devices also provides for two situations in which the notified body is required to seek scientific advice from a national competent authority for medicinal products or from the EMA before proceeding with certification: - When the (non-viable) substances of human origin used to manufacture a medical device can be considered as a medicinal product within the meaning of the Directive 2001/83/EC and their action is ancillary to that of the device (Annex IX-5.2(b) of the Regulation on medical devices). The notified body may not issue a CE certificate where the competent authority consulted in the field of medicinal products has issued a negative opinion device (Annex IX-5.2(e) of the Regulation on medical devices);
- When the (non-viable) substances of human origin used to manufacture a medical device are intended to be absorbed by or locally dispersed in the human body (Annex IX-5.4(b) of the Regulation on medical devices). The opinion of the consulted authority must be taken into account by the notified body, although a negative opinion does not necessarily result in a prohibition on issuing the CE certificate. (Annex IX-5.4(d) of the Regulation on medical devices).
|
Conformity assessment based on type examination (Annex X) and on product conformity verification (Annex XI)
Assessment based on type examination (Annex X):
This assessment involves an examination of a representative sample of the production (i.e. a type) and a review of the technical documentation to ensure compliance with the regulation. A representative sample of the production is provided by the manufacturer to a NB. The NB evaluates it to verify that the manufactured medical device conforms to the technical documentation (Annex X-3(c) and (g) of the Regulation on medical devices)
Following this evaluation, and if the device complies with the regulation, the NB issues an EU type-examination certificate. (Annex X-4 of the Regulation on medical devices)
It should be noted that additional specific procedures applicable within the framework of conformity assessment based on a quality management system and the assessment of technical documentation (Annex X-6 by reference to Annex IX-5 of the Regulation on medical devices) mentioned above are also applicable to the following medical devices:
- Class III implantable medical devices
- Medical devices manufactured using human tissues or cells
Conformity assessment based on product conformity verification (Annex XI):
After obtaining the EU type-examination certificate, the manufacturer may choose between two options for the subsequent steps of the conformity assessment procedure for its medical device.
The first option consists in an assessment of the Quality Management System very similar to that described above (Annex IX of the Regulation on medical devices), except that it is aimed at demonstrating conformity with the type/sample assessed and certified in the preceding stage, as well as with the related documentation. Conformity allows the NB to issue an EU quality assurance certificate. (Annex XI, Part A of the Regulation on medical devices)
The second option is more burdensome and more constraining. It requires each manufactured medical device to undergo examinations and testing directly by the NB (or by a laboratory designated by it), in order to ensure each one’s compliance with the regulation and conformity with the type/sample assessed in the preceding stage. Compliance enables the notified body to issue an EU product verification certificate. (Annex XI, Part B of the Regulation on medical devices)
The certificated issued by the NB are valid for the specified period and for a maximum of 5 years. At the manufacturer’s request, the validity of the certificate may be extended by up to five years each time, following a new conformity assessment. (Article 56(2) of the Regulation on medical devices)
Last steps before placing on the market:
Regardless of the procedure chosen, once the EU certificates have been obtained, the manufacturer is still required to:
- Draw up an EU declaration of conformity (Article 10.6 of the Regulation on medical devices)
- Affix the CE marking of conformity on its medical device (Article 10.6 of the Regulation on medical devices)
- Register in the dedicated databases through the EUDAMED platform by providing the information listed in Annex VI to the MDR, or verify such information where registration has already been completed. (Article 29 of the Regulation on medical devices)
4. Identification and preparation of the required documentation
In order to develop a medical device that complies with regulatory requirements, the manufacturer can refer to several documents related to documentation.
On the one hand, the manufacturer must establish and continuously update the technical documentation provided for in Annex II of the Regulation on medical devices, as well as the post-market surveillance documentation set out in Annex III of the Regulation on medical devices.
On the other hand, additional standards may be applied during the design and development or manufacturing phases. They mainly consist in relevant ISO standards.
Their application is voluntary, and facilitates the demonstration of conformity.
Among ISO standards relevant for medical devices using tissues and cells of human origin, the following can be cited:
There is also a list of references to harmonised standards under Regulation (EU) 2017/745 on medical devices, published in the Official Journal of the European Union.
This list is regularly updated on the European Commission’s website here.
5. Requirements for placing a medical device on the market
Requirements applicable to all medical devices (Annex I of the Regulation on medical devices)
To be placed on the market or put into service, a medical device must comply with general safety and performance requirements. (Annex I of the Regulation on medical devices)
They include:
The general requirements concern the safety and effectiveness of medical devices with regard to the performance and intended purpose specified by the manufacturer: medical devices must be designed and manufactured in such a way as to be suitable for their intended purpose under normal conditions of use; risks must be reduced as far as possible, and the benefit-risk balance must be favourable.
These requirements include the implementation of a risk management system and its systematic and periodic updating throughout the entire life cycle of medical devices. They must be complied with throughout the entire life cycle of medical devices, including during storage and transport, so as to prevent any alteration of their performance and characteristics. (Chapter I of the Annex I of the Regulation on medical devices)
Requirements regarding design and manufacture concern the reduction of the risks associated with chemical (toxic, carcinogenic, mutagenic, endocrine disruptors) or biological substances (microbial infections), physical factors (risk of injury, radiation, etc.), and with electronic systems and software (active and software-based medical devices). (Chapter II of the Annex I of the Regulation on medical devices)
Requirements regarding the information supplied with the device concern labelling and the instructions for use. (Chapter III of the Annex I of the Regulation on medical devices)
Specific requirements related to the nature of the medical deviceMore specifically, additional requirements apply to medical devices manufactured from non-viable cells and tissues of human origin (Annex I, Chapter II, Section 13.1 of the Regulation on medical devices) until 6 August 2027: - Donation, procurement and testing of tissues and cells must comply with Directive 2004/23/EC.
- Safety of the medical device must be ensured, especially with regard to potential pathogens. To this end, appropriate sourcing methods and validated methods for the removal or inactivation of such pathogens must be applied during manufacturing.
- The use of tissues and cells must comply with the traceability requirements laid down in Directives 2004/23/EC and 2002/98/EC.
As from 7 August 2027: Where non-viable substances of human origin or their derivatives incorporate, as an integral part, a medical device: - And their action is principal to that of the medical device: the SoHO regulation applies to both these non-viable substances of human origin (and their derivative) and to the final combination. OR
- And their action is ancillary to that of the medical device: the SoHO regulation applies to these non-viable substances of human origin (and their derivative) with regard to donor registration, donor history review and medical examination, donor screening, collection, processing, quality control, storage, release, and distribution, for the purpose of their incorporation into the medical device. But the Regulation on medical devices applies to the final combination. (Article 2.8 of the SoHO Regulation)
Where substances of human origin are collected for the manufacture of medical devices, the SoHO Regulation applies to donor registration, donor history review and medical examination, donor screening, collection, and release activities relating to substances of human origin. (Article 2.6 of the SoHO Regulation) If activities relating to the storage, distribution, importation or exportation of substances of human origin are carried out, the SoHO Regulation shall apply up to and including their distribution to a medical device manufacturer. (Article 2.6 of the SoHO Regulation) However, where substances of human origin are used for the manufacture of medical devices intended for autologous use (i.e. where the donor IS also the recipient), the SoHO Regulation applies only to activities relating to the “screening of SoHO donors or persons from whom SoHO are collected for autologous or intra-partner use” and to the collection of substances of human origin. (Article 2.7 of the SoHO Regulation) Finally, the Annex I of the Directive 2001/83/EC relating to medicinal products must serve as the basis for the notified body’s verification of the quality and safety of non-viable substances of human origin when: These requirement laid down in Annex I of the Directive 2001/83/EC therefore also apply to the manufacturer in relation to those substances. |
6. Registration obligations applicable to all economic operators
Before the marketing of medical devices, and even prior to the conformity assessment procedure, economic operators (manufacturers, authorised representatives and importers) are required to register. To do so, they may choose to register once on the EUDAMED platform by providing the information listed in Annex VI, Part A-1 of the Regulation on medical devices, registration that will be effective throughout the EU. (Article 30 and 31 of the Regulation on medical devices)
However, since the platform is not yet fully operational (Article 123(3)(d) of the Regulation on medical devices), direct registration with national competent authorities remains possible, although such registration is valid only in the territory of the Member State where the relevant formalities were carried out.
From 28 May 2026, the registration on EUDAMED will be mandatory for all economic operators. (Commission Decision (EU) 2025/2371 and Regulation (EU) 2024/1860)
In addition to registration, the obligations of each economic operator are specified in the chapter II of the Regulation on medical devices.
Only the manufacturer’s obligations will be detailed in the dedicated subsection.