1. Déterminer la nature du produit
1 Article 2(1)(d) of the Regulation (EC) No 1394/2007 on ATMPs
2 Article 1(10) §1 of the Regulation on medical devices
2. Déterminer la classe du dispositif médical
La règlementation sur les dispositifs médicaux s’articule autour d’une classification des dispositifs médicaux fondée sur le risque, des dispositifs les moins à risque, de classe I, aux dispositifs les plus à risque, de classe III. Les conditions de mise sur le marché sont donc de plus en plus strictes au fur et à mesure que la classe, et donc le risque, augmente.
La classification d’un dispositif médical est donc centrale puisqu’elle déterminera les exigences applicables.
3. Identifier et choisir la procédure d'évaluation
Pour pouvoir apposer un marquage CE sur un dispositif médical, condition obligatoire pour la mise sur le marché (Article 20 du Règlement sur les dispositifs médicaux), un dispositif médical doit faire l’objet d’une des procédures d’évaluation de la conformité (Article 52 du Règlement sur les dispositifs médicaux). Cette évaluation, réalisée par un organisme notifié pour tous les dispositifs médicaux autres que ceux de classe I, a pour objectif de s’assurer du respect des exigences réglementaires par le fabricant.
Et comme déjà évoqué, la classe d’un dispositif médical est déterminante pour définir les exigences qui lui seront applicables. Il en va donc de même pour la procédure d’évaluation de la conformité.
Pour les dispositifs médicaux de classe III (tous les dispositifs médicaux fabriqués à partir de substances d’origine humaine non-viables) :
L’évaluation de la conformité du dispositif médical exigera l’intervention d’un organisme notifié. Pour cela, le fabricant est libre de son choix et peut introduire sa demande d’évaluation auprès de n’importe quel organisme notifié, désigné par une autorité nationale responsable des organismes notifiés, au sein de l’UE (voir Liste de organismes notifiés – en anglais ou Actors and Network database).
Une seule condition est imposée pour guider le choix de l’organisme notifié par le fabricant: « à condition que l'organisme notifié choisi soit désigné pour réaliser les activités d'évaluation de la conformité liées aux types de dispositifs concernés ». (Article 53(1) du Règlement sur les dispositifs médicaux)
Avant d’introduire une demande d’évaluation auprès d'un organisme notifié, le fabricant d’un dispositif médical de classe III doit lui attribuer un identifiant unique (IUD-ID) et l’enregistrer. (Article 29.3 du Règlement sur les dispositifs médicaux)
Le Règlement d’exécution (UE) 2026/977 établit certaines exigences uniformes en matière de gestion de la qualité et de procédures applicables aux activités d’évaluation de la conformité exécutées par un organisme notifié (principalement devis/coût, délais et recertification).
Deux options s’offrent ensuite au fabricant concernant l’évaluation par un ON :
- Une évaluation de la conformité sur la base d'un système de gestion de la qualité et de l'évaluation de la documentation technique (Annexe IX du Règlement sur les dispositifs médicaux)
- Une évaluation sur la base de l’examen de type (Annexe X) et de la vérification de la conformité du produit (Annexe XI)
Une évaluation de la conformité sur la base d'un système de gestion de la qualité et de l'évaluation de la documentation technique (Annexe IX du Règlement sur les dispositifs médicaux)
Cette évaluation comprend à la fois une évaluation du système de gestion de la qualité et une évaluation de la documentation technique.
Evaluation du système de gestion de la qualité par l’organisme notifié (ON) choisi :
Après soumission par le fabricant d’une demande d’évaluation du système de gestion de la qualité, l’ON évalue les objectifs qualité du fabricant, les mesures mises en place, l’organisation et les qualifications des personnels, les systèmes de contrôle et de surveillance de la conception jusqu’à la production des dispositifs. Dans le cadre de cette évaluation, l’ON effectue un audit sur place, chez le fabricant et ses fournisseurs et/ou sous-traitants le cas échéant, afin de contrôler le Système de Gestion de la Qualité (SGQ) et sa mise en œuvre effective.
À l’issue de cette évaluation et si le SGQ est effectivement conforme à la réglementation, l’ON délivre un certificat UE relatif au système de gestion de la qualité, première étape vers l’obtention du marquage de conformité CE. (Annexe IX-2 du Règlement sur les dispositifs médicaux)
Evaluation de la documentation technique par l’organisme notifié (ON) choisi :
Après soumission par le fabricant d’une demande d’évaluation de la documentation technique, l’ON effectue également un contrôle approfondi de la documentation technique décrite dans les Obligations applicables au fabricant de dispositifs médicaux . Celui-ci consistera notamment en l’évaluation des preuves cliniques apportées par le fabricant, du rapport bénéfice/risque, et de l’équivalence avec un dispositif médical déjà existant le cas échéant, en tenant compte du caractère innovant du dispositif médical, du plan de gestion des risques, du public visé, du plan de surveillance après commercialisation et du suivi clinique après commercialisation (voir Obligations applicables au fabricant de dispositifs médicaux). (Annexe IX-4.1 à 4.8 du Règlement sur les dispositifs médicaux)
À l’issue de cette évaluation, et si le dispositif est conforme à la réglementation, l’ON délivre cette fois un certificat d'évaluation UE de la documentation technique. (Annexe IX-4.9 du Règlement sur les dispositifs médicaux)
Il faut noter qu’il existe des procédures spéciales complémentaires pour certains dispositifs médicaux :
Plus particulièrement, pour les dispositifs médicaux fabriqués à partir de tissus ou cellules non-viables d’origine humaine (donc de classe III) : Avant de prendre sa décision, l’ON doit solliciter une des autorités compétentes en matière de tissus et cellules humaines au sens de la directive 2004/23/CE / autorité SoHO selon le Règlement (UE) 2024/1938. En s’appuyant sur le résumé de l’évaluation préliminaire de la documentation technique contenant les informations pertinentes relatives à ces tissus et cellules, l’autorité compétente sollicitée rendra un avis sur les différents aspects concernant les substances d’origine humaine. En cas d’avis défavorable de la part de l’autorité compétente, l’ON ne peut délivrer de certificat de conformité. En cas d’avis favorable, l’ON conserve la possibilité de délivrer ou non un tel certificat, à la condition toutefois de prendre en compte les opinions exprimées dans ledit avis et de l’intégrer à sa documentation concernant le dispositif médical. L’ON a également obligation de communiquer sa décision finale à l’autorité compétente ainsi sollicitée. (Annexe IX-5.3.1 du Règlement sur les dispositifs médicaux)
Le Règlement sur les dispositifs médicaux prévoit également deux cas où l’organisme notifié est dans l’obligation de solliciter l’avis scientifique d’une autorité nationale compétente pour les médicaments ou de l’EMA avant de procéder à la certification : |
Une évaluation de la conformité sur la base de l’examen de type (Annexe X) et de la vérification de la conformité du produit (Annexe XI)
Évaluation sur la base de l’examen de type (Annexe X) :
Cette évaluation comprend un examen d’un échantillon représentatif de la production (un type) et un examen de la documentation technique pour s’assurer de sa conformité avec la réglementation. Un échantillon représentatif de la production est mis à disposition d’un ON par le fabricant. L’ON l’évalue afin de vérifier que le dispositif médical produit correspond bien à la documentation technique (Annexe X-1 et 3(b) du Règlement sur les dispositifs médicaux). Pour cela, l’ON réalise lui-même ou charge un laboratoire indépendant de réaliser des essais sur l’échantillon. (Annexe X-3(c), (f) et (g) du Règlement sur les dispositifs médicaux)
À l’issue de cette évaluation, et si le dispositif est conforme à la réglementation, l’ON délivre un certificat d’examen UE de type. (Annexe X-4 du Règlement sur les dispositifs médicaux)
À noter que les procédures spéciales complémentaires applicables dans le cadre de l’évaluation de la conformité sur la base d'un système de gestion de la qualité et de l'évaluation de la documentation technique (Annexe X-6 par renvoi à l’Annexe IX-5 du Règlement sur les dispositifs médicaux) mentionnées ci-dessus sont également applicables aux dispositifs médicaux suivants :
- Dispositifs médicaux de classe III implantables
- Dispositifs médicaux fabriqués à partir de tissus ou cellules humaines
Évaluation de la conformité sur la base de la vérification de la conformité du produit (Annexe XI) :
Après l’obtention du certificat d’examen UE de type, le fabricant peut choisir entre deux options quant à la suite de la procédure de conformité de son dispositif médical.
La première option consiste en une évaluation du Système de Gestion de la Qualité très comparable à celle décrite précédemment (Annexe IX du Règlement sur les dispositifs médicaux), à ceci près qu’elle vise à la conformité avec le type/l’échantillon évalué et certifié lors de la phase précédente et la documentation afférente. La conformité permet la délivrance par l’ON d’un certificat UE d'assurance de la qualité. (Annexe XI, Partie A du Règlement sur les dispositifs médicaux)
La seconde option est plus lourde et contraignante. Elle nécessite que chaque dispositif médical produit fasse l’objet d’examens et de tests directement par l’ON (ou un laboratoire désigné par lui) afin de s’assurer que chaque dispositif médical respecte la réglementation et correspond au type/échantillon évalué lors de la phase précédente. La conformité permet la délivrance par l’ON d’un certificat UE vérification du produit. (Annexe XI, Partie B du Règlement sur les dispositifs médicaux)
Les certificats délivrés par les ON sont valables pendant la période indiquée et pour une durée maximale de 5 ans. À la demande du fabricant, la durée de validité du certificat peut être prolongée d'une durée maximale de cinq ans à chaque fois, à la suite d’une nouvelle évaluation de la conformité (Article 56(2) du Règlement sur les dispositifs médicaux).
Dernières étapes avant la mise sur le marché:
Quelle que soit la procédure choisie, une fois obtenus les certificats UE, le fabricant doit encore :
4. Identifier et établir la documentation requise
Afin de développer un dispositif médical conforme aux exigences règlementaires, le fabricant peut s’appuyer sur plusieurs textes relatifs à la documentation.
D’une part, le fabricant doit obligatoirement établir et mettre à jour la documentation technique prévue à l’Annexe II du Règlement sur les dispositifs médicaux, et la documentation technique relative à la surveillance après commercialisation prévue à l’annexe III du Règlement sur les dispositifs médicaux.
D’autre part, des normes additionnelles peuvent être appliquées pendant la conception et le développement, ou la fabrication. Il s’agit principalement des normes ISO pertinentes.
Leur application est volontaire, et elle facilite les démonstrations de conformité.
Parmi les normes ISO pertinentes aux dispositifs médicaux basés sur des cellules et tissus d’origine humaine, on peut citer :
Il existe aussi une liste de références des normes harmonisées relatives au règlement 2017/745 sur les dispositifs médicaux, et publiées au Journal Officiel de l’Union européenne.
Cette liste est régulièrement mise à jour sur le site de la Commission européenne ici.
5. Exigences à respecter pour mettre dispositif médical sur le marché
Pour être mis sur le marché ou mis en service, un dispositif médical doit être conforme aux exigences générales en matière de sécurité et de performances. (Annexe I du Règlement sur les dispositifs médicaux)
Elles comprennent :
Les exigences générales concernent la sécurité et l’efficacité des dispositifs médicaux au regard des performances et de la destination prévues par le fabricant : la conception et la fabrication des dispositifs médicaux doivent être adaptées à leur destination dans des conditions normales d’utilisation ; les risques doivent être réduits autant que possible, et la balance bénéfice-risque doit être positive.
Ces exigences incluent la mise en place d’un système de gestion des risques et sa mise à jour systématique et périodique pour l’ensemble du cycle de vie des dispositifs médicaux. Elles doivent être respectées pendant toute la durée de vie des dispositifs médicaux, y compris pendant le stockage et le transport afin d’éviter l’altération de leurs performances et caractéristiques. (Chapitre I de l’Annexe I du Règlement sur les dispositifs médicaux)
Les exigences relatives à la conception et à la fabrication concernent la réduction des risques liés aux substances chimiques (toxiques, cancérogènes, mutagènes, perturbateurs endocriniens) ou biologiques (infections microbiennes), aux propriétés physiques (risque de blessure, rayonnements, etc.) et aux systèmes électroniques et logiciels (dispositifs actifs et dispositifs logiciels). (Chapitre II de l’Annexe I du Règlement sur les dispositifs médicaux)
Les exigences relatives aux informations fournies avec le dispositif concernent l’étiquetage et la notice d’utilisation. (Chapitre III de l’Annexe I du Règlement sur les dispositifs médicaux).
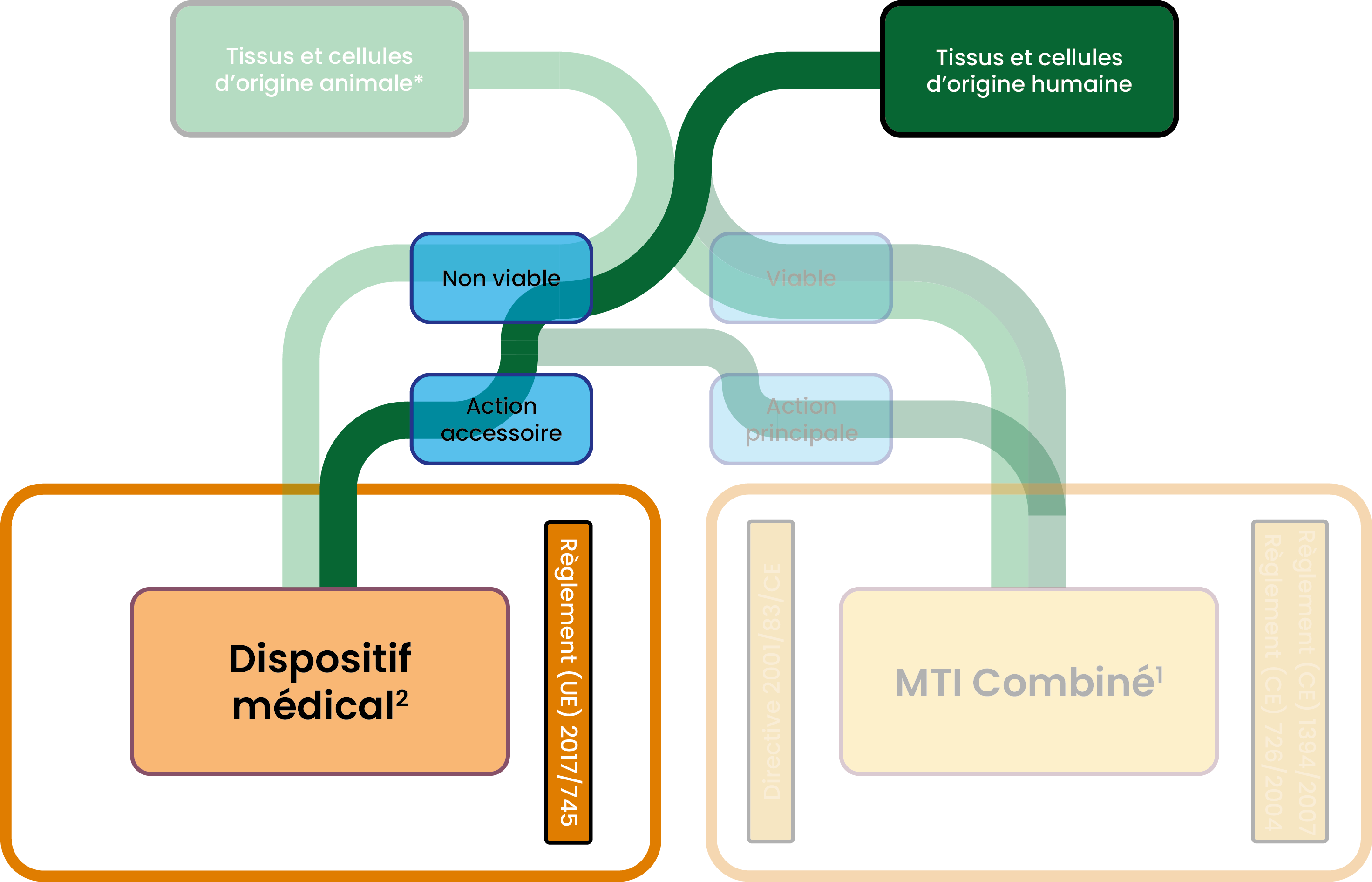
Exigences particulières liées à la nature du dispositif médicalConcernant plus particulièrement les dispositifs médicaux fabriqués à partir de cellules et tissus non-viables d’origine humaine, d’autres exigences s’appliquent (Annexe I, Chapitre II, Section 13.1 du Règlement sur les dispositifs médicaux) jusqu’au 6 août 2027: - le don, l'obtention et le contrôle des tissus et cellules doivent être conformes à la Directive 2004/23/CE.
- La sécurité des dispositifs médicaux doit être garantie, en particulier en ce qui concerne d’éventuels agents pathogènes. Pour cela, des méthodes d'approvisionnement appropriées et des méthodes validées d’élimination ou inactivation de tels agents doivent être appliquées au cours de la fabrication.
- L’utilisation des tissus et cellules doit respecter les obligations de traçabilité issues des Directives 2004/23/CE et 2002/98/CE.
A partir du 7 Août 2027 : Lorsque des substances d’origine humaine non viables ou leurs dérivés incorporent comme partie intégrante un dispositif médical : - Et qu’elles ont une action essentielle par rapport au dispositif médical : le règlement SoHO s’applique à la fois à ces substances d’origine humaine non viables (ou à leurs dérivés) et à la combinaison finale. OU
- Et qu’elles ont une action accessoire par rapport au dispositif médical : le règlement SoHO s’applique à ces substances d’origine humaine non viables (ou à leurs dérivés) pour l’enregistrement des donneurs, la vérification des antécédents des donneurs et leur examen médical, le contrôle des donneurs, le prélèvement, la transformation, le contrôle de la qualité, le stockage, la libération, et la distribution de ces substances en vue de leur intégration dans le dispositif médical. Mais le Règlement sur les dispositifs médicaux s’applique à la combinaison finale. (Article 2.8 du Règlement SoHO)
Lorsque des substances d’origine humaine sont prélevées pour fabriquer des dispositifs médicaux, le règlement SoHO s’applique aux activités d’enregistrement des donneurs, de vérification des antécédents des donneurs et leur examen médical, de contrôle des donneurs, de prélèvement et de libération des substances d’origine humaine. (Article 2.6 du Règlement SoHO). Si des activités de stockage, distribution, importation et exportation sont menées sur des substances d’origine humaine, le règlement SoHO doit être respecté jusqu’à leur distribution à un fabricant de dispositifs médicaux incluse. (Article 2.6 du Règlement SoHO). Cependant, lorsque des substances d’origine humaine sont utilisées pour fabriquer des dispositifs médicaux destinés à un usage autologue (le donneur EST aussi le receveur), le règlement SoHO ne s’applique qu’aux activités de « contrôle des donneurs de SoHO ou des personnes auprès desquelles des SoHO sont prélevées pour un usage autologue ou une utilisation intra-relationnelle » et de prélèvement des substances d’origine humaine. (Article 2.7 du Règlement SoHO) Enfin, l'Annexe I de la Directive 2001/83/CE concernant les médicaments doit servir de base à la vérification par l’organisme notifié de la qualité et la sécurité des substances d’origine humaines non-viables lorsque : Ces exigences résultant de l’Annexe I de la Directive 2001/83/CE s’imposent donc également au fabricant pour ce qui concerne ces substances. |
6. Obligations d'enregistrement pour tous les opérateurs économiques
Avant la commercialisation et avant même la procédure de conformité des dispositifs médicaux, les opérateurs économiques (fabricants, mandataires et importateurs) doivent s’enregistrer. Pour cela, ils peuvent choisir de s’enregistrer de manière unique sur la plateforme EUDAMED en renseignant les informations listées à l’annexe VI, Partie A-1 du Règlement sur les dispositifs médicaux, enregistrement qui sera effectif pour l’ensemble de l’UE. (Article 30 et 31 du Règlement sur les dispositifs médicaux)
Cependant, la plateforme n’étant pas pleinement opérationnelle (Article 123(3)(d) du Règlement sur les dispositifs médicaux), l’enregistrement directement auprès des autorités compétentes nationales demeure possible, celui-ci ne valant toutefois que pour le territoire de l’État membre où les démarches ont été effectuées.
À partir du 28 Mai 2026, l’enregistrement sur EUDAMED sera obligatoire pour tous les opérateurs économiques. (Commission Decision (EU) 2025/2371 and Regulation (EU) 2024/1860)
Au-delà de l’enregistrement, les obligations de chaque opérateur économique sont précisées dans le chapitre II du Règlement sur les dispositifs médicaux.
Nous détaillons dans la sous-entrée dédiée les obligations du fabricant seulement.