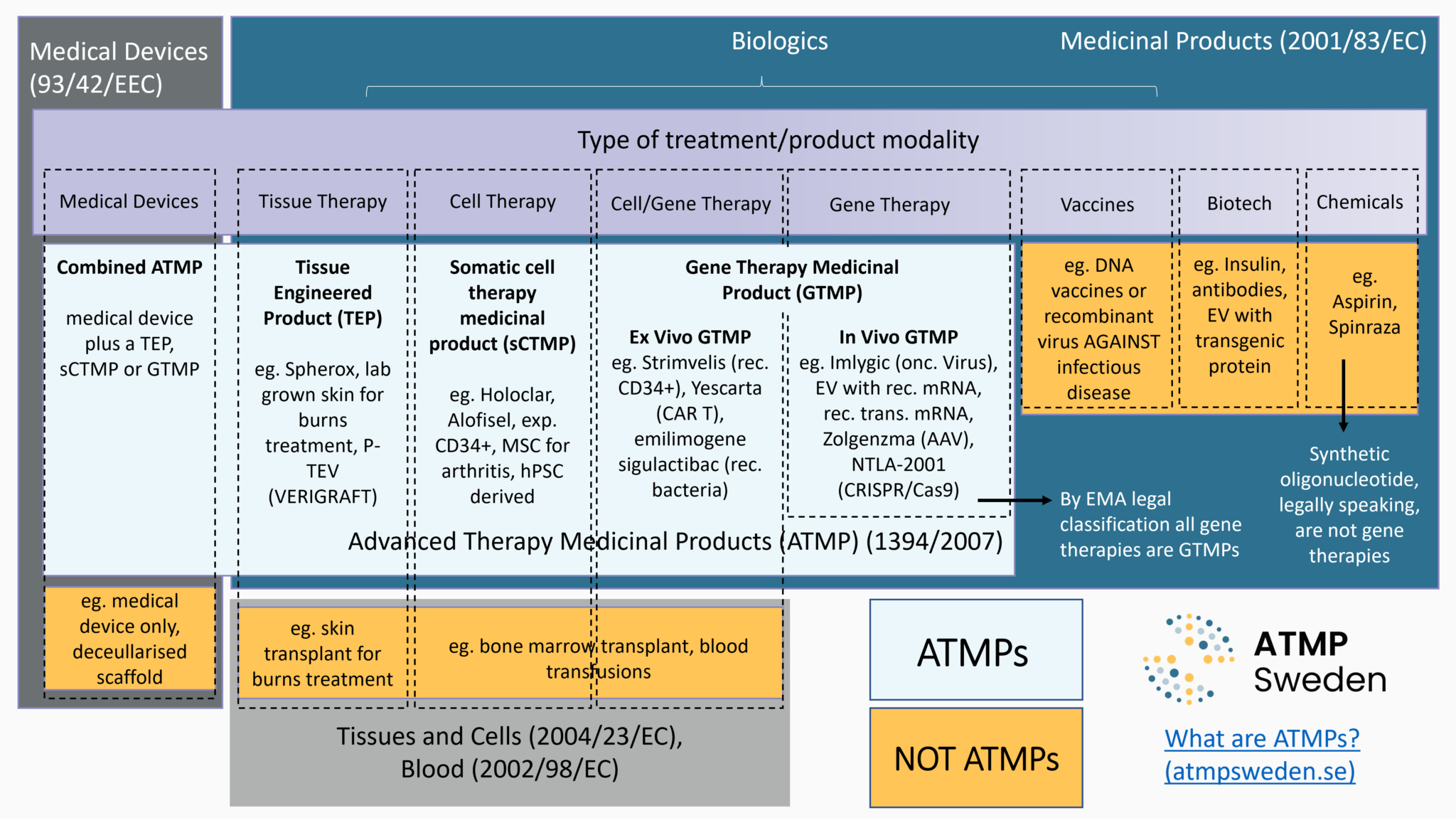
La classification des thérapies détermine les mécanismes juridiques qui régissent la réglementation des thérapies géniques et cellulaires afin de garantir la qualité, la sécurité et l'efficacité de ces thérapies sur le marché de l'UE.
La première étape des aspects réglementaires concernant le développement des thérapies est de définir le produit en cours de développement, à savoir déterminer sa classification juridique. S'agit-il d'un médicament, d'un dispositif médical, ou de cellules ou tissus utilisés à des fins thérapeutiques ? Si c'est un médicament, s'agit-il d'un Médicament de Thérapie Innovante (MTI), d'un médicament orphelin et/ou d'un médicament à usage pédiatrique ? La classification juridique détermine le cadre réglementaire applicable, c'est-à-dire les exigences ultérieures que le produit doit satisfaire, ainsi que les procédures pertinentes menant à la commercialisation, telles que l'Autorisation de Mise sur le Marché (AMM) pour les MTI. Voir aussi : Commercialisation.
Le droit (principalement le droit de l'Union européenne, mais aussi les droits nationaux) donne des définitions légales pour chaque type de produit. Différents critères ont été établis pour distinguer le type de thérapie génique et cellulaire en développement et, par conséquent, quel ensemble de réglementations s'applique à la fois pour l'accès au marché et pour les décisions de remboursement. Les critères principaux sont :
- L'échelle de développement du produit :
- Le produit est-il développé au niveau « industriel » ou non ?
- Les degrés de manipulation des tissus et cellules :
- Les tissus et cellules ont-ils été manipulés de manière substantielle ou non ?
- Les tissus et cellules sont-ils utilisés pour les mêmes fonctions essentielles chez le receveur que chez le donneur ?
Malgré ces définitions légales et ces critères, il reste difficile de savoir exactement quel type de thérapie génique et cellulaire est en développement selon le droit. Il est recommandé aux développeurs de contacter les autorités réglementaires au niveau national ou européen ainsi que les organismes de remboursement dès que possible.
Le Comité des Thérapies Innovantes (CAT) de l'Agence européenne des médicaments (EMA) est particulièrement impliqué dans la classification juridique des MTI dans l'Union européenne. Conformément au règlement (CE) n° 1394/2007 sur les MTI, l'EMA a mis en place une procédure de classification des MTI pour aider les développeurs à clarifier si leur produit est ou non un MTI, et quel cadre réglementaire s'applique. Le CAT au sein de l'EMA émet des recommandations scientifiques basées sur la conformité du produit aux critères scientifiques et règlementaires définissant les MTI. Les résumés de ces recommandations, y compris les justifications fournies, sont publiés sur le site de l’EMA : voir Recommandations scientifiques sur la classification des médicaments de thérapie innovante (en anglais uniquement).
Les MTI sont un type de thérapie génique et cellulaire, dont le développement est particulièrement soutenu par le droit de l'Union européenne, à la fois dans des objectifs de santé publique (niveau élevé de protection de la santé humaine dans l'UE) et économiques (bon fonctionnement du marché intérieur et compétitivité). Il existe plusieurs réglementations pour l'accès des patients aux thérapies géniques et cellulaires, y compris lorsqu’il s’agit de MTI. Les MTI relèvent du cadre juridique des médicaments, en particulier des médicaments biologiques. Les MTI inclut quatre sous-types de médicaments basés sur des gènes, des cellules ou des tissus : Médicaments de thérapie génique (MTG), Médicament de thérapie cellulaire somatique (MTCs), Produit issu de l'ingénierie tissulaire (PIT), Médicament combiné de thérapie innovante. Selon le droit de l'UE :
- Médicament de thérapie génique (MTG):
« un médicament biologique qui a les caractéristiques suivantes: a) il contient une substance active qui contient ou constitue un acide nucléique recombinant administré à des personnes en vue de réguler, de réparer, de remplacer, d’ajouter ou de supprimer une séquence génétique; b) son effet thérapeutique, prophylactique ou diagnostique dépend directement de la séquence d’acide nucléique recombinant qu’il contient ou au produit de l’expression génétique de cette séquence. Les vaccins contre les maladies infectieuses ne sont pas compris dans les médicaments de thérapie génique. » [Directive 2001/83/CE, Annexe I, Partie IV, modifiée par la directive de la Commission 2009/120/CE] - Médicament de thérapie cellulaire somatique (PTCs):
« un médicament biologique qui présente les caractéristiques suivantes: a) contient ou consiste en des cellules ou des tissus qui ont fait l’objet d’une manipulation substantielle de façon à modifier leurs caractéristiques biologiques, leurs fonctions physiologiques ou leurs propriétés structurelles par rapport à l’usage clinique prévu, ou des cellules ou tissus qui ne sont pas destinés à être utilisés pour la ou les mêmes fonctions essentielles chez le receveur et le donneur ; b) est présenté comme possédant des propriétés permettant de traiter, de prévenir ou de diagnostiquer une maladie à travers l’action métabolique, immunologique ou pharmacologique de ses cellules ou tissus, ou est utilisé chez une personne ou administré à une personne dans une telle perspective. »
[Directive 2001/83/CE, Annexe I, Partie IV, modifiée par la directive de la Commission 2009/120/CE]- Note : L'annexe I du règlement (CE) n° 1394/2007 sur les MTI énumère une liste non exhaustive de manipulations non substantielles : « découpage, broyage, façonnage, centrifugation, trempage dans des solutions antibiotiques ou antimicrobiennes, stérilisation, irradiation, séparation, concentration ou purification de cellules, filtration, lyophilisation, congélation, cryoconservation, vitrification. »
- Produit issu de l'ingénierie tissulaire (PIT) :
« un produit qui contient des cellules ou tissus issus de l’ingénierie cellulaire ou tissulaire, ou en est constitué, et - qui est présenté comme possédant des propriétés lui permettant de régénérer, réparer ou remplacer un tissu humain, ou est utilisé chez l’être humain ou administré à celui-ci dans ce but. » [Article 2(1)(b) du règlement (CE) n° 1394/2007]
- Médicament combiné de thérapie innovante :
est un médicament de thérapie génique, un médicament de thérapie cellulaire somatique ou un produit issu de l'ingénierie tissulaire associé à un dispositif médical. Il « doit incorporer comme partie intégrante un ou plusieurs dispositifs médicaux (…), ou bien un ou plusieurs dispositifs médicaux implantables actifs (…), et sa partie cellulaire ou tissulaire doit contenir des cellules ou des tissus viables, ou sa partie cellulaire ou tissulaire contenant des cellules ou des tissus non viables doit être susceptible d’avoir sur le corps humain une action qui peut être considérée comme essentielle par rapport à celle des dispositifs précités » [Article 2(1)(d) du règlement (CE) n° 1394/2007]
Dans cette section, nous fournirons des informations concernant les différentes classifications juridiques des thérapies géniques et cellulaires et des voies réglementaires qui leur sont associées en vue de l’utilisation clinique de ces thérapies.
Remerciements
Publication : 29/01/2022
Dernière misa à jour : 24/05/2025
Auteur :
Hsin-Yu Kuo, EuroGCT Project Manager - Research Information and Networks
Relecture :
Aurélie Mahalatchimy, EuroGCT WP4 Convenor, UMR 7318 DICE CERIC, Aix-Marseille University, CNRS, Aix-en-Provence- France
Traduction de l'anglais au français en mai 2025 :
Victoria Burakova-Lorgnier, Juriste, UMR 7318 DICE CERIC, Aix-Marseille Université, Aix-en-Provence, France
Aurélie Mahalatchimy