Soutien de plusieurs autorités compétentes en faveur du développement de médicaments innovants
Introduction
Des procédures d’interaction précoce avec les autorités compétentes sont mises en œuvre afin de soutenir le développement des médicaments, en particulier des médicaments innovants, et de relever les défis spécifiques auxquels ils sont confrontés. Ce soutien prend la forme de services et de procédures réglementaires permettant aux développeurs de médicaments d’interagir avec les autorités compétentes grâce à des mécanismes existants tout au long du développement d’un médicament. Un contact précoce avec les autorités compétentes est essentiel pour les développeurs lorsqu’ils travaillent à la mise sur le marché d’un nouveau médicament au sein de l’Union européenne. Dans cette optique, de nombreux services et procédures ont été mis en place par les autorités compétentes afin de faciliter le dialogue et la circulation des informations sur les questions réglementaires, ainsi que pour prendre en compte les différents cadres juridiques existants.
D’une part, un contact précoce peut être établi avec une seule autorité compétente, soit au niveau de l’Union européenne, soit au niveau national, via ces procédures ou dispositifs. D’autre part, certains dispositifs sont conçus pour permettre aux développeurs d’entrer en contact simultanément avec plusieurs autorités compétentes. Lorsque plusieurs autorités sont impliquées, les interactions peuvent être multiniveaux au sein de l’Union européenne. Par exemple, avec l’Agence européenne des médicaments (EMA) et une ou plusieurs autorités nationales compétentes (telles que les organismes d’évaluation des technologies de santé (ETS) dans l’Union européenne ou les agences nationales du médicament).
Ces procédures peuvent également cibler une étape spécifique du calendrier de recherche et développement, comme les essais cliniques (par exemple : conseils préalables à une demande d’essai clinique – Pre-CTA – ou projets pilotes SAWP-CTCG). Elles peuvent aussi concerner deux autorités situées dans différentes régions du monde, notamment l’EMA pour le marché de l’Union européenne et l’Agence américaine des produits alimentaires et médicamenteux (Food and Drug Administration- FDA) pour le marché des États-Unis. Les interactions précoces peuvent également avoir lieu aux niveaux nationaux avec plusieurs autorités compétentes (par exemple dans le cadre du conseil scientifique national simultané).
Six procédures peuvent être considérées comme impliquant plusieurs autorités compétentes :
Avis scientifique du Scientific Advice Working Party (SAWP) – projet pilote du Clinical Trials Coordination Group (CTCG) visant à interagir avec toutes les agences nationales des médicaments participantes par l’intermédiaire de l’Agence européenne des médicaments,sur la pertinence d’un plan d’essai clinique pour soutenir une demande d’essai clinique (CTA – Clinical Trial Application) dans un État membre ou pour accompagner les développeurs dans leur demande d’autorisation de mise sur le marché (AMM – Marketing Authorisation Application) ;
Le projet pilote Pre-CTA est coordonné par le CTCG afin d’interagir avec toutes les agences nationales des médicaments participantes sur les questions réglementaires relatives au dossier de demande d’essai clinique (CTA – Clinical Trial Application) avant la soumission d’une demande d’essai clinique dans leClinical Trials Information System (CTIS) ;
Avis scientifique national simultané (SNSA – Simultaneous National Scientific Advice) visant à interagir avec deux ou plusieurs services d’innovation des agences nationales des médicaments de l’Espace économique européen (EEE) ;
Avis scientifique parallèle visant à interagir à la fois avec l’Agence européenne des médicaments (EMA) et la FDA ;
Consultations scientifiques conjointes visant à interagir avec tous les organismes d’évaluation des technologies de santé (ETS – Health Technology Assessment, HTA) de l’Union européenne via le groupe de coordination ETS (HTA Coordination Group) ;
Consultations scientifiques conjointes parallèles visant à interagir avec plusieurs organismes d’ETS (HTA bodies) de l’Union européenne et avec l’Agence européenne des médicaments.
Acteurs principaux
ACT-EU : ACT-EU a été créé par une action conjointe de l’Agence européenne des médicaments (EMA), du réseau des Responsables des agences du médicament (HMA) et de la Commission européenne (DG SANTE et DG RTD). Il a été établi en janvier 2022 dans le contexte de la mise en œuvre du nouveau règlement relatif aux essais cliniques (CTR). ACT-EU dispose d’un programme de travail comportant 11 domaines prioritaires. Pour en savoir plus sur ACT-EU sur leur site web. Le programme de travail 2023-2026 ainsi que le dernier programme annuel 2025-2026 y sont disponibles. Le programme de travail ACT-EU 2025–2026 explique clairement que l’objectif de cette action est d’accélérer les essais cliniques dans l’Union européenne en soutenant des essais plus efficaces grâce à l'utilisation de processus multiples (innovation, réglementation et technologie) afin de favoriser l’innovation. « La vision est de disposer d’essais cliniques meilleurs, plus rapides et optimisés, au bénéfice des patients et des systèmes de santé en Europe. Une coordination fluide entre les parties prenantes, les autorités réglementaires et les comités d’éthique renforce la collaboration transfrontalière. La plateforme multipartite ACT-EU est centrale à cet égard. »
Groupe de coordination des essais cliniques (CTCG) : Le CTCG est un groupe de travail du réseau des Responsables des agences du médicament (HMA). Il est composé d’experts des agences nationales des médicaments chargés de l’évaluation et de la supervision des essais cliniques. Ce groupe vise à renforcer l’attractivité de l’Union européenne / de l’Espace économique européen (UE/EEE) pour les essais cliniques en promouvant l’harmonisation et l’optimisation de la réglementation. En savoir plus sur ce groupe ici. Pour consulter le mandat du CTCG, suivez ce lien.
EU Innovation Network (EU-IN) : Groupe de travail créé en 2015 pour soutenir le système européen de réglementation des médicaments (EMRN) dans le développement de nouveaux médicaments innovants. Il agit sous mandat conjoint de l’Agence européenne des médicaments (EMA) et du réseau des Responsables des agences du médicament (HMA), et inclut des représentants des services nationaux d’innovation. Il vise à faciliter le développement de produits innovants aux niveaux nationaux et européen, notamment en favorisant l’utilisation des dispositifs réglementaires existants tels que les avis scientifiques. Il a été conçu pour renforcer la collaboration entre les agences nationales des médicaments et l’EMA sur les questions réglementaires liées aux thérapies émergentes et aux technologies associées. ( EU Innovation Network (EU-IN) | European Medicines Agency (EMA)). Ce groupe mène également des activités de « veille technologiques » (horizon scanning) afin d’identifier les nouvelles tendances émergentes dans le domaine des médicaments. Un sous-groupe dédié est chargé du dispositif d’avis scientifique national simultané (SNSA), piloté par le Paul Ehrlich Institute (PEI), l’agence nationale des médicaments en Allemagne. Ce sous-groupe met en œuvre des procédures SNSA depuis 2020. L’EU-IN est également chargé de promouvoir la participation des autorités compétentes aux projets financés par des sources externes. (Voir, Considerations for research / project teams seeking competent authority participation in externally funded regulatory science and public health research projects related to medicinal products, 2023 EMA/38599/2023, 19 janvier, (en anglais)).
Agence européenne des médicaments (EMA) : L’Agence européenne des médicaments est une agence de l’Union européenne dont l’objectif est de protéger et promouvoir la santé humaine et animale. Elle est responsable de l’évaluation scientifique des médicaments de thérapie innovante destinés à être commercialisés dans l’Union européenne et l’Espace économique européen. L’Agence propose un certain nombre d’incitations et de procédures pour soutenir les développeurs de médicaments et améliorer l’accès des patients aux traitements.
Réseau européen de réglementation des médicaments (EMRN) : Le réseau européen de réglementation des médicaments est un système réglementaire régional coordonné dans lequel la Commission européenne, l’Agence européenne des médicaments et les autorités de réglementation des médicaments des 30 pays de l’EEE collaborent. Le réseau garantit que les médicaments autorisés sont sûrs, efficaces et de haute qualité. Des informations complémentaires sont disponibles sur le site de l’Agence Européenne des Médicaments (EMA), dans la rubrique 'European medicines regulatory network'.
Responsables des agences du médicament (HMA) : Le réseau des Responsables des médicaments est composé des directeurs des agences nationales des médicaments. Ces agences sont responsables, dans leur pays respectif, de la réglementation des médicaments à usage humain et vétérinaire dans l’Espace économique européen. Le HMA coopère avec l’Agence européenne des médicaments et la Commission européenne pour renforcer la coopération européenne en matière d’activités réglementaires. Pour plus d’information sur l’institution ou sur ses missions veuillez suivre le lien suivant.
Organismes en charge de l’évaluation des technologies de la santé (HTA bodies): Organismes chargés d’évaluer les technologies de santé (telles qu’un médicament, un dispositif médical ou une procédure médicale ou chirurgicale). L’évaluation fournie comprend des informations synthétiques sur les aspects d’ordre médical, social et relatifs aux patients ainsi que sur les questions économiques et éthiques liés à l’utilisation d’une technologie de santé et ses conséquences. (Voir la définition de l’évaluation des technologies de la santé et des technologies de santé dans le glossaire d’EuroGCT)
Groupe de coordination des États membres sur l’évaluation des technologies de la santé (HTACG) (ci-après «groupe de coordination») : Groupe composé de représentants des États membres de l’Union européenne (principalement issus des autorités et organismes nationaux d’évaluation des technologies de santé), établi par le règlement de l’UE concernant l’évaluation des technologies de la santé. Ses principales missions sont de coordonner et d’adopter les travaux conjoints d’HTA réalisés par ses sous-groupes dans le cadre de ce règlement, ainsi que d’adopter des documents d’orientation méthodologiques et procéduraux. Le HTACG vise également à assurer la coopération entre les organismes pertinents de l’Union européenne (par exemple l’Agence européenne des médicaments et les organismes HTA), ainsi que l’implication appropriée des parties prenantes et des experts dans ses travaux. (pour en savoir plus sur le groupe de coordination HTA). Le HTACG est notamment responsable des consultations scientifiques conjointes.
Services d’innovation : Les services d’innovation sont des guichets, bureaux ou services établis par les agences nationales des médicaments. Ils centralisent les procédures disponibles pour les développeurs de produits de santé innovants (notamment les médicaments innovants), y compris ceux visant à faciliter les interactions précoces avec les autorités réglementaires. Leur objectif est d’aider les développeurs à s’orienter parmi les différents dispositifs de soutien et à choisir celui qui correspond le mieux à leurs besoins. Les services d’innovation constituent un moyen de cibler et de répondre aux besoins des développeurs, et de leur fournir le soutien nécessaire pour mettre sur le marché de nouveaux médicaments innovants. Des exemples de dispositifs et de procédures pour lesquels les services d’innovation apportent des orientations incluent : la procédure d’avis scientifique au niveau national, ou encore la procédure d’avis scientifique national simultané (SNSA) lorsqu’elle est disponible. Il est important de souligner que tous les pays de l’Espace économique européen (EEE) ne disposent pas d’une entité dédiée de ce type, pour accompagner les développeurs dans l’obtention des informations nécessaires au développement d’un nouveau médicament. La liste des Services d’innovation dans l’Espace Économique Européen (EEE) est disponible dans la base de données EuroGCT Acteurs et Réseaux.
Développeurs de médicaments : Toute entité ou personne morale ayant l’objectif de mettre des médicaments à disposition des patients. Les développeurs de médicaments sont les principaux bénéficiaires des interactions précoces avec les autorités réglementaires, telles que les agences nationales des médicaments, l’Agence européenne des médicaments, ainsi que les organismes HTA. Les développeurs de médicaments peuvent être des PME, des institutions académiques, des grandes entreprises pharmaceutiques ou des organisations à but non lucratif.
Agences nationales des médicaments : Au niveau national, les agences nationales des médicaments sont les autorités réglementaires chargées de promouvoir la santé humaine et de garantir la sécurité des médicaments. Il s’agit des organismes publics chargés de veiller au respect de la réglementation des médicaments, placés sous l’autorité du ministère de la Santé dans la plupart des pays. Les agences nationales du médicament évaluent, autorisent, réglementent et contrôlent les médicaments à usage humain ainsi que les produits de santé au niveau de l’État. Une liste complète des autorités compétentes nationales dans les États membres de l’Union européenne est disponible dans la base de données EuroGCT Acteurs et Réseaux.
Autorités réglementaires : Entités désignées par la loi pour l’autorisation, l’approbation, l’évaluation, la supervision, la surveillance et/ou l’élaboration de lignes directrices dans un domaine spécifique. Dans le contexte des thérapies géniques et cellulaires, plusieurs autorités réglementaires interviennent tout au long du cycle de développement : agences du médicament (européennes ou nationales), organismes d’évaluation des technologies de santé, autorités de financement et d’autorisation de la recherche, offices des brevets, instances/comités d’éthique, organismes notifiés (dans le cadre des combinaisons avec des dispositifs médicaux), autorités en charge des données, etc. Veuillez noter que, dans la plupart des documents de l’Agence européenne des médicaments (EMA), le terme « regulators » est utilisé pour désigner uniquement l’EMA et les agences nationales du médicament. (D’après le glossaire des termes d’EuroGCT)
Groupe de travail sur le conseil scientifique (SAWP, de l’anglais Scientific Advice Working Party) : Groupe de travail de l’Agence européenne des médicaments (EMA), mis en place par le comité des médicaments à usage humain (CHMP), chargé de fournir des avis scientifiques et une assistance pour les protocoles aux développeurs. Il constitue un acteur majeur des interactions précoces entre l’EMA et les parties prenantes. Le SAWP participe aux procédures d’avis scientifique parallèle avec l’EMA et la FDA, ainsi qu’au projet pilote SAWP-CTCG dans le cadre de l’initiative ACT-EU. Pour chaque demande d’avis scientifique ou d’assistance au protocole, deux délégués sont désignés comme coordinateurs de l’évaluation, et au moins un comme relecteur de l’avis final (voir Human Medicines Division Mandate, objectives and rules of procedure of the Scientific Advice Working Party (SAWP), 14 avril 2025, EMEA/CHMP/SAWP/69686/04 Rev 19). Pour en savoir plus sur le SAWP, consultez le site de l’European Medicines Agency (EMA).
Acteur en recherche d’interaction : Le développeur de médicaments souhaitant recourir à une procédure ou un service spécifique destiné à favoriser les interactions précoces entre autorités compétentes et parties prenantes. L’objectif de ces interactions est souvent de faciliter le développement de futurs médicaments commercialisables.
Food and Drug Administration (FDA) : La Food and Drug Administration est l’agence nationale des médicaments des États-Unis. Elle est responsable de la protection et de la promotion de la santé publique, notamment par l’évaluation et l’autorisation des médicaments et dispositifs médicaux destinés à être commercialisés aux États-Unis.
Définitions
Parcours adaptatifs : Les parcours adaptatifs (Adaptive Pathways) faisaient partie de la stratégie de l’Agence européenne des médicaments (EMA) visant à favoriser un accès rapide aux médicaments pour les patients dans le besoin. Cette procédure réglementaire a été conçue pour permettre un accès progressif et adapté des patients aux médicaments. Elle s’appuyait sur d’autres procédures réglementaires existantes, telles que le conseil scientifique. Le projet pilote relatif aux parcours adaptatifs s’est déroulé de 2014 à 2016. Il a offert une plateforme permettant aux développeurs d’engager des discussions informelles avec l’EMA et d’autres parties prenantes, notamment des patients et des organismes d’évaluation des technologies de santé (HTA bodies), afin d’examiner des questions scientifiques et réglementaires. Ces procédures ont aidé les développeurs à formuler leurs questions et les ont orientés vers d’autres formes d’interaction précoce si nécessaire, telles que le conseil scientifique. Elles ont été remplacées par une autre procédure d’interaction précoce : le conseil scientifique parallèle avec les organismes ETS (HTA bodies), qui a évolué en consultations scientifiques communes parallèles (consultations scientifiques communes menées parallèlement à l’obtention de l’avis scientifique de l’EMA) suite à l’adoption du Règlement ETS en 2021. Pour plus d’informations, voir les parcours de développement
Interaction précoce : L’interaction précoce correspond à la possibilité de contacts entre les développeurs de médicaments et les autorités compétentes, via des procédures ou services dédiés, à un stade précoce du développement d’un médicament (principalement en phase de recherche et développement). Les procédures ou services varient en fonction du type de produit, du statut du développeur et des besoins identifiés. Les interactions précoces sont généralement des dispositifs volontaires auxquels les développeurs peuvent choisir de recourir ; aucun n’est obligatoire.
Médicament innovant : Un médicament innovant est un médicament contenant une substance active ou une combinaison de substances actives qui n’a pas encore été autorisée. (D’après le glossaire des termes de régulation de l’EMA)
Consultation scientifique commune (JSC) : La consultation scientifique commune (JSC) est une procédure volontaire permettant à un développeur de médicaments d’obtenir des orientations individuelles concernant les informations, données, analyses et autres éléments de preuve susceptibles d’être requis à partir des essais cliniques par les autorités nationales compétentes chargées d’évaluer et/ou de décider du remboursement des médicaments par les systèmes nationaux d’assurance maladie. Cette procédure est conduite au niveau européen via le groupe de coordination ETS (HTA CG), composé de représentants des États membres issus des autorités et organismes ETS (HTA bodies). Elle peut faciliter la future procédure obligatoire d’évaluation clinique conjointe (JCA) des médicaments autorisés au niveau de l’Union européenne, dont les résultats devront être dûment pris en compte par les autorités nationales compétentes lors des décisions de remboursement.
Consultation scientifique commune parallèle avec l’EMA et les organismes ETS : Procédure volontaire permettant aux développeurs de médicaments d’obtenir des retours à la fois de l’EMA et des organismes ETS (HTA bodies), par l’intermédiaire du groupe de coordination HTA au niveau de l’Union européenne (HTA CG), sur leurs plans de génération de données probantes, tant pour l’autorisation de mise sur le marché que pour le remboursement des médicaments. Cette procédure vise à surmonter les défis liés à la coexistence de différentes activités réglementées (autorisation de mise sur le marché et remboursement des médicaments), avec des prises de décision à des niveaux distincts : au niveau de l’Union européenne pour les décisions d’autorisation de mise sur le marché des médicaments de thérapie innovante (MTI), et au niveau national pour les décisions de remboursement. Cette procédure aide les développeurs à générer simultanément des données solides destinées à différentes autorités compétentes, dans le cadre des différentes étapes réglementaires nécessaires pour mettre les MTI à disposition des patients. En effet, l’EMA est chargée de l’évaluation scientifique des MTI en vue de leur autorisation de mise sur le marché, tandis que les organismes d’ETS (HTA bodies), représentés au sein du groupe de coordination ETS au niveau de l’Union européenne (HTA CG), sont responsables de l’évaluation en vue des décisions de remboursement. La procédure de consultation scientifique commune parallèle s’appuie sur des initiatives pilotes et des collaborations antérieures entre agences du médicament et organismes d’ETS (HTA bodies), menées notamment par l’EMA, EUnetHTA et la Commission Européenne. Celles-ci incluent le rapport du projet pilote sur les avis scientifiques parallèles entre autorités réglementaires et organismes d’ETS, l’initiative de dialogue précoce d’EUnetHTA, le projet SEED (2008–2013), financé par l’Union européenne ainsi que des travaux conjoints sur le niveau d’alignement entre autorités réglementaires du médicament et organismes d’ETS dans le cadre des avis parallèles. Pour plus d’informations, voir : Partenaires et réseaux : organismes d’évaluation des technologies de santé.
Conseil scientifique parallèle EMA–FDA : Procédure optionnelle et volontaire permettant aux développeurs de médicaments de bénéficier des conseils à la fois de l’EMA dans l’Union européenne et de la FDA aux États-Unis. Elle est généralement utilisée au cours de la phase de recherche et développement du cycle de vie du produit. L’avis scientifique parallèle EMA-FDA permet aux développeurs de produits complexes de gagner un temps précieux lorsqu’ils envisagent de déposer des demandes d’autorisation de mise sur le marché à la fois aux États-Unis et dans l’Union européenne. En effet, ils peuvent bénéficier des conseils de plusieurs autorités compétentes situées dans différentes régions, impliquant des cadres réglementaires distincts à respecter et à appliquer.
Conseil scientifique du SAWP, Groupe de Coordination des Essais Cliniques (SAWP–CTCG) : La justification de la mise en place des projets pilotes consolidés sur les essais cliniques, y compris le pilote SAWP-CTCG, est de proposer une procédure visant à favoriser la mise en œuvre d’essais cliniques de meilleure qualité, plus rapides et optimisés au sein de l’Union européenne. Ce projet pilote a pour objectif de fournir et de renforcer les conseils relatifs aux aspects scientifiques des essais cliniques ainsi qu’aux programmes de développement, dans le cadre du réseau européen de réglementation des médicaments (European Medicines Regulatory Network – EMRN). Il vise à accompagner les développeurs dans la conduite des essais cliniques ainsi que dans la préparation des demandes d’autorisation de mise sur le marché. Cela est rendu possible grâce à la collaboration entre le groupe de travail sur les avis scientifiques de l’Agence Européenne des Médicaments (SAWP) et le Clinical Trials Coordination Group (CTCG), impliquant les États membres et leurs agences nationales du médicament.
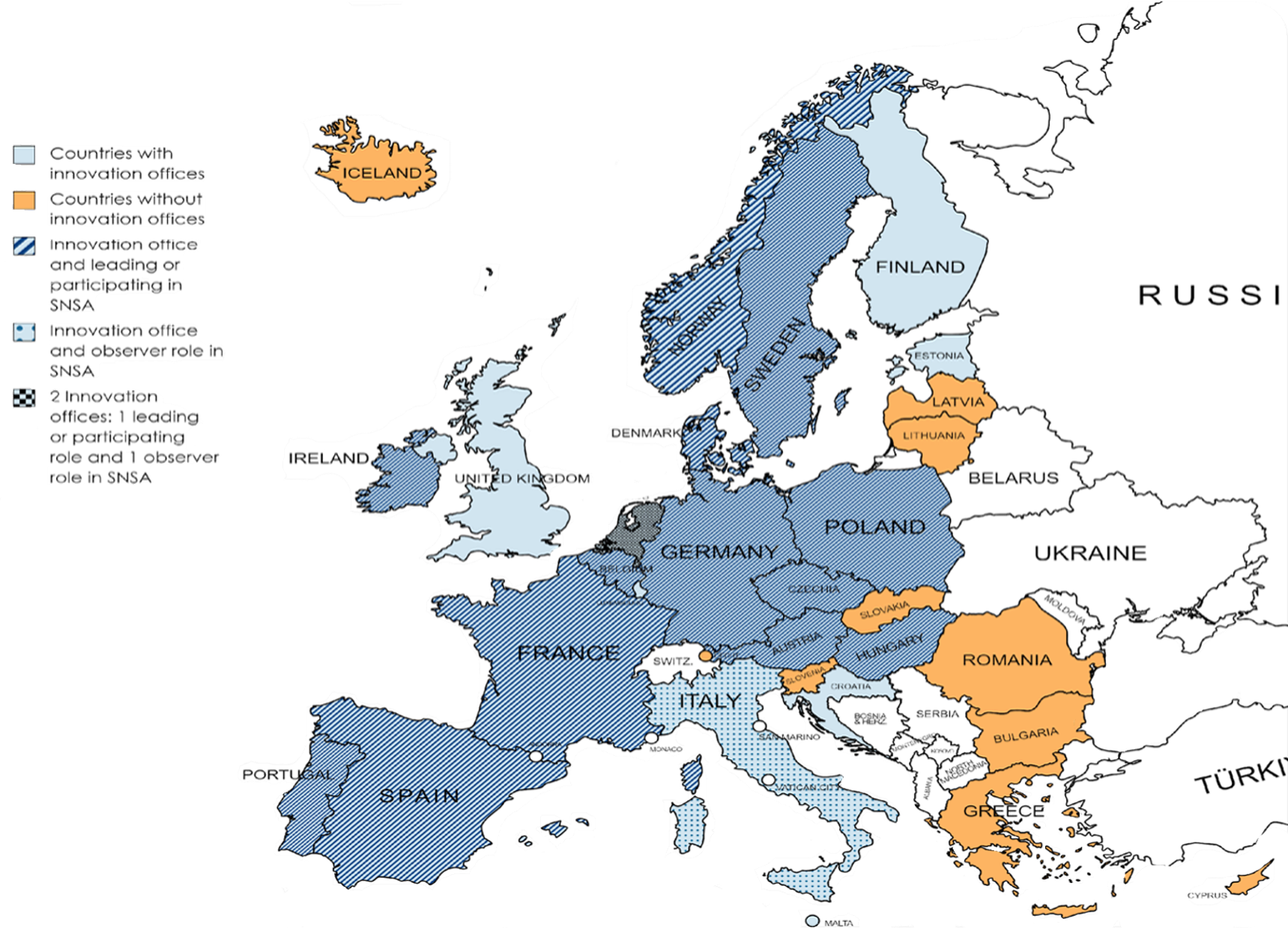
Avis scientifique national simultané (SNSA): Procédure pilotée par les HMA et l’EMA, destinée aux développeurs souhaitant obtenir des avis scientifiques auprès de plusieurs autorités nationales compétentes simultanément. Initialement lancée en février 2020 en tant que projet pilote visant à améliorer l’efficacité en termes de délais et de coûts pour les développeurs, cette procédure est entrée dans une phase d’optimisation en 2025. Elle permet aux développeurs d’entrer en contact avec deux (ou davantage, si justifié) autorités nationales compétentes issues de différents pays européens, et de solliciter des avis scientifiques ou réglementaires au niveau national, via leurs services d’innovation respectifs, à condition que ces autorités participent au dispositif. Le SNSA constitue un outil complémentaire aux dispositifs existants, notamment aux avis scientifiques délivrés par l’EMA uniquement ainsi que les conseils réglementaires fournis aux niveaux nationaux dans chaque pays. Les pays participants ainsi que les services d’innovation dans les pays de l’Espace économique européen (EEE) sont présentés dans la cartographie ci-dessous. Cette cartographie met en évidence le fait que les services d’innovation sont souvent associés à la participation au SNSA, bien que certains pays disposant d’un tel service n’y participent pas (par exemple : l’Estonie, la Finlande, la Croatie et le Royaume-Uni). Toutefois, tous les pays participant au SNSA disposent d’un service d’innovation, ce qui souligne le rôle essentiel de ces structures dans la mise en œuvre de cette procédure.
Source : A. Delage, L.-S. Gilbert, A. Mahalatchimy, Mapping regulators’ early interactions procedures to support innovation, Congrès de la European Society for Gene & Cell Therapy (ESGCT) 2023, Bruxelles, Belgique, 24–27 octobre 2023, poster.
STARS : Action de coordination et de soutien (CSA) financée par l’Union européenne, intitulée Strengthening Training of Academia in Regulatory Science (STARS), menée du 1er janvier 2019 au 30 juin 2022. Cette initiative visait à soutenir le développement de médicaments innovants au sein du milieu académique en lui apportant le soutien réglementaire nécessaire. Elle avait pour objectifs de s’adresser aux innovateurs académiques dans le domaine du médicament, de combler le déficit de connaissances en matière de réglementation et de renforcer le dialogue entre le monde académique et les autorités réglementaires.
La coordination des différentes exigences émanant de plusieurs autorités réglementaires, ainsi que l’identification des interlocuteurs compétents auxquels adresser des questions spécifiques, peuvent constituer un obstacle au développement d’un médicament. Toutefois, les dispositifs impliquant plusieurs autorités réglementaires permettent de surmonter ces difficultés.
Pour les développeurs souhaitant concevoir un nouveau médicament, connaître, comprendre et respecter les nombreuses exigences réglementaires aux niveaux nationaux et européen représente un défi majeur. Ils doivent naviguer entre différents cadres juridiques, et comprendre comment les articuler entre eux.
Par ailleurs, les développeurs peuvent chercher à accéder à différents marchés à l’échelle mondiale, où les règles varient d’un territoire à l’autre. Par exemple, les exigences pour accéder aux marchés de l’Union européenne et des États-Unis sont différentes. Ainsi, un développeur souhaitant intégrer ces deux marchés peut être amené à engager des interactions précoces avec les autorités compétentes « régionales » afin de comprendre les exigences scientifiques et réglementaires applicables. Dans cette optique, l’EMA et la FDA ont mis en place une forme d’avis scientifique conjoint appelée « Conseil scientifique parallèle EMA–FDA», portant sur des questions scientifiques.
Les autorités compétentes contribuent également à combler le déficit de connaissances des développeurs en mettant en place de nouveaux dispositifs et procédures destinés à faciliter la mise à disposition de médicaments innovants pour les patients. Ces différentes initiatives peuvent évoluer rapidement en raison du dynamisme du domaine et de l’expérience acquise. Il peut donc être difficile de connaître, comprendre et suivre l’évolution des différentes procédures proposées par les autorités compétentes à différents niveaux (national, européen et international).
En outre, la terminologie et les descriptions de certaines procédures peuvent prêter à confusion. Pendant longtemps, l’expression « avis parallèle » désignait deux types de procédures : celle entre l’EMA et la FDA, et celle entre l’EMA et les ETS, ce qui a généré une confusion inutile. Depuis l’entrée en vigueur du règlement européen sur les ETS, la terminologie et les procédures ont été clarifiées. La procédure permettant d’interagir avec plusieurs organismes d’ETS est désormais appelée consultations scientifiques communes (Joint Scientific Consultations – JSC). Il reste toutefois possible de combiner les consultations de l’EMA et des organismes d’ETS via des consultations scientifiques communes parallèles. Par ailleurs, la procédure permettant d’interagir à la fois avec l’EMA et la FDA est désignée comme Conseil scientifique parallèle avec les États-Unis ou avec la FDA. Malgré les efforts de clarification engagés par le règlement ETS et poursuivis par l’EMA, le terme « parallèle » demeure utilisé pour désigner deux procédures distinctes, bien qu’il implique systématiquement la participation de l’EMA aux côtés d’une autre autorité réglementaire.
Enfin, le coût de ces procédures peut constituer un obstacle, même si certaines sont gratuites et que des incitations existent pour les PME et le secteur académique, en fonction des autorités compétentes impliquées et de la procédure visée.
Opportunités et incitations
Les autorités compétentes, ou autorités réglementaires, mettent en place des services et des procédures visant à faciliter l’obtention de réponses de plusieurs régulateurs par les développeurs de médicaments. L’objectif principal est de soutenir le développement des médicaments et leur accès aux patients. Un objectif implicite est également d’éviter que des recherches innovantes ne soient freinées par la complexité des parcours réglementaires, notamment en raison des différences de compétences et des cadres législatifs applicables.
On observe une forte volonté de favoriser la collaboration entre les différentes autorités, certains pays disposant déjà d’une expérience en la matière à travers des accords bilatéraux et des procédures spécifiques.
Certaines initiatives internationales portées par les agences réglementaires permettent l’émergence de nouveaux programmes, favorisant ainsi de nouvelles collaborations et, à terme, le développement et l’accès à de nouveaux médicaments. Par exemple, la FDA étudie actuellement un nouveau programme pilote intitulé Gene Therapies Global Pilot (CoGenT), qui devrait initialement inclure les membres réglementaires de l’International Council for Harmonisation (ICH), notamment l’Union européenne, les États-Unis, le Japon, le Canada et la Suisse. Ce programme repose sur une collaboration entre les membres réglementaires permanents de l’ICH. Les partenaires peuvent participer à des réunions réglementaires internes ou à des réunions avec les développeurs ayant sollicité une interaction. Ce projet pilote est mentionné dans le rapport annuel 2024 (CY 2024 report) du directeur de la FDA (pour lire le rapport, voir ci-joint). Il vise à promouvoir une convergence réglementaire à l’échelle internationale. Bien qu’il s’agisse encore d’un projet à l’état de développement, il illustre l’importance accordée par plusieurs pays à la coopération en matière réglementaire et à l’harmonisation des exigences entre les différents marchés, afin de faciliter l’accès des médicaments innovants aux patients.
Il est essentiel que les parties prenantes soient informées de l’existence de ces procédures et de leur évolution. De nombreux événements sont organisés par les autorités réglementaires afin de sensibiliser les acteurs concernés. Certains sont recensés dans une veille juridique régulière sur le site d’ELSIBI. On constate un réel intérêt et une volonté de renforcer l’accompagnement des parties prenantes en facilitant leurs interactions avec les autorités compétentes, notamment par la création d’espaces d’échange. Cette dynamique incite les autorités à développer de nouvelles procédures et à clarifier les exigences nécessaires pour accéder au marché ou maximiser les chances d’y accéder. À titre d’exemple, la mise en place de services d’innovation constitue un indicateur fort de l’engagement de nombreux pays et autorités compétentes en faveur du développement de médicaments innovants. Par ailleurs, une volonté d’harmonisation des approches se dessine, notamment par l’identification de défis communs à l’échelle régionale et la mise en place de procédures partagées. La procédure d’avis scientifique national simultané (SNSA) en est une illustration : elle répond à des problématiques similaires rencontrées par plusieurs autorités nationales, nécessitant des données comparables.
Enfin, d’autres dispositifs de soutien existent, notamment sous forme de collaborations entre pays via des accords spécifiques. C’est le cas de l’accord BENELUXA réunissant la Belgique, l’Autriche, l’Irlande, le Luxembourg et les Pays-Bas (pour en savoir plus, voir le site Beneluxa). Ce partenariat vise à garantir un accès durable aux médicaments innovants à des coûts abordables, en mutualisant les efforts de plusieurs pays. L’objectif est de faciliter le dialogue et l’échange d’informations sur les politiques du médicament, avec l’espoir d’avoir un impact positif sur les coûts et les stratégies associées. Il permet également aux pays de négocier conjointement les prix avec l’industrie, renforçant ainsi la transparence entre les États en matière de tarification des médicaments.
Étapes pratiques
Les développeurs peuvent entrer en contact avec leurs agences nationales du médicament, mais également avec les Responsables des agences du médicament (HMA) et ses groupes de travail, le groupe de coordination HTA et les organismes ETS, l’EMA et/ou la FDA en fonction de leur objectif et du type d’interaction qu’ils doivent engager afin de commercialiser leur produit.
Six procédures différentes coexistent actuellement et permettent aux développeurs d’interagir directement avec plusieurs autorités réglementaires :
Si l’interaction recherchée concerne les essais cliniques et que le développeur souhaite obtenir un avis scientifique à la fois sur les exigences relatives aux demandes d’essais cliniques et à l’autorisation de mise sur le marché, il devrait envisager de solliciter un avis via le piloted’avis scientifique SAWP CTCG.
Si l’interaction recherchée concerne un développeur souhaitant obtenir un conseil réglementaire sur le dossier de demande d’essai clinique (CTA) avant de soumettre une demande d’essai clinique dans CTIS, il peut recourir au pilote d’avis Pre-CTA.
Si l’interaction recherchée concerne des essais cliniques devant être menés dans plus d’un État membre de l’UE, les développeurs de médicaments (souvent le promoteur, voir essais cliniques) devraient envisager d’entrer dans une procédure d’avis scientifique national simultané (SNSA) afin d’interagir avec plusieurs services d’innovation de différentes agences nationales du médicament dans l’Espace économique européen.
Si l’interaction recherchée concerne la génération de données probantes pour l’autorisation de mise sur le marché d’un MTI à la fois sur les marchés de l’UE et des États-Unis, le développeur devrait envisager d’initier une procédure de Conseil scientifique parallèle EMA-FDA.
Si l’interaction recherchée concerne le remboursement d’un MTI (ou plus largement d’une technologie de santé) dans plusieurs États membres de l’UE, le développeur devrait envisager d’utiliser la procédure de consultation scientifique commune afin d’interagir avec l’ensemble des organismes d’ETS (HTA bodies) de l’Union européenne via le groupe de coordination ETS (HTA CG).
Si l’interaction recherchée concerne la génération de données probantes à la fois pour l’autorisation de mise sur le marché au niveau de l’UE et pour le remboursement d’un MTI dans plusieurs États membres de l’UE, le développeur devrait envisager d’initier une procédure de consultation scientifique commune parallèle afin d’interagir avec le groupe de coordination ETS (HTA CG) et avec l’EMA.
Veuillez trouver ci-dessous plus de détails sur les étapes pratiques de chacune de ces procédures.
Projets pilotes d’avis consolidés sur les essais cliniques
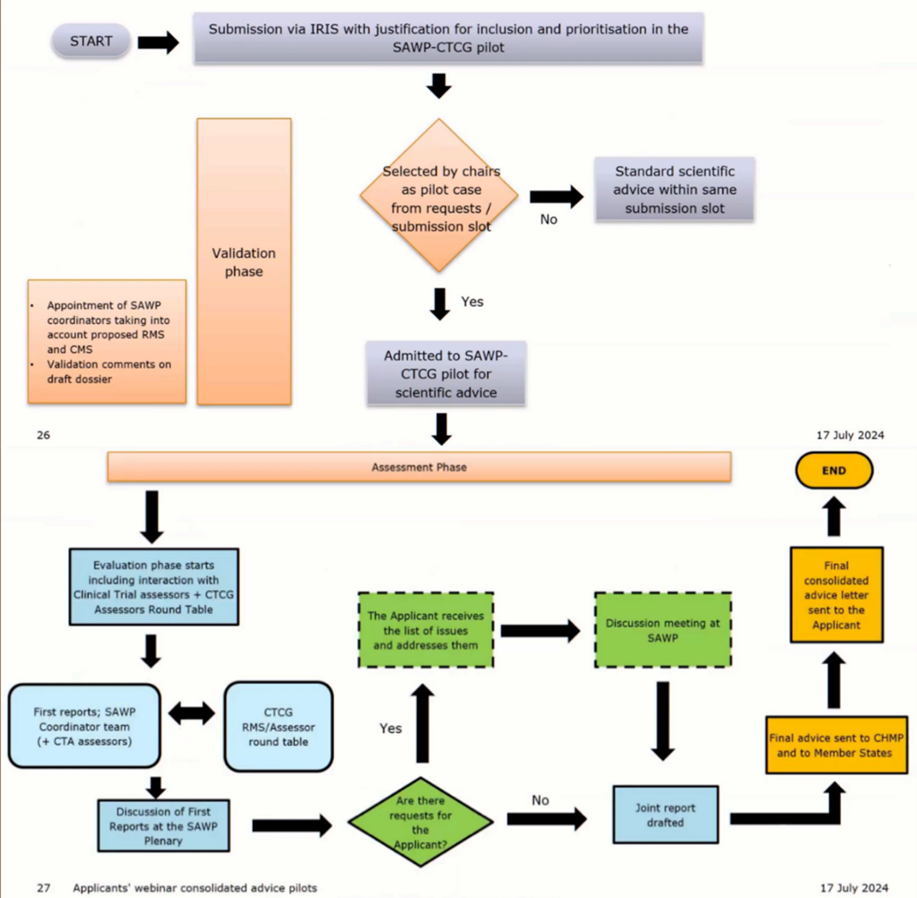
1. Conseil scientifique du projet pilote SAWP CTCG
Le pilote SAWP CTCG est un type d’avis scientifique portant sur l’adéquation du design d’un essai clinique pour soutenir une demande d’autorisation de mise sur le marché (AMM) au niveau européen lorsque la procédure est centralisée et/ou des demandes d’essais cliniques (CTA) auprès d’une agence nationale du médicament dans un État membre spécifique. Le pilote vise à créer une vision et des avis harmonisés sur les designs d’essais cliniques pour les deux types de demandes (CTA et AMM) à travers les États membres de l’UE. Il repose sur une collaboration renforcée entre le groupe de travail sur les avis scientifiques de l’EMA (SAWP) et les membres du groupe de coordination des essais cliniques des États membres (CTCG).
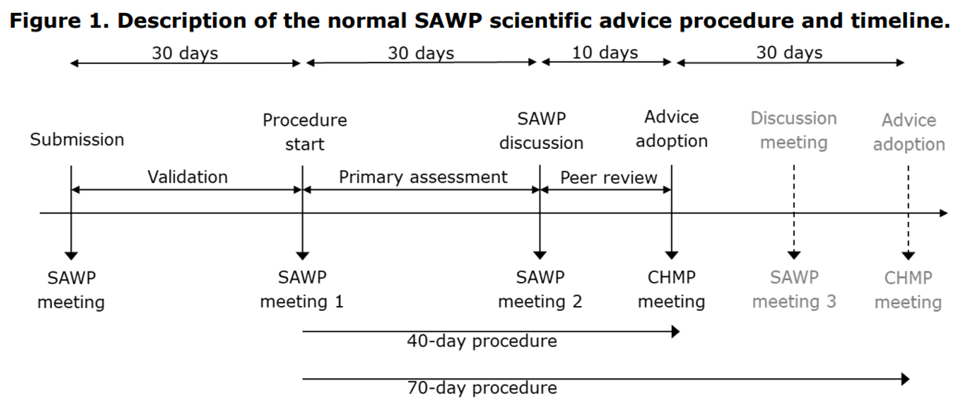
La procédure standard et le calendrier des avis scientifiques du SAWP s’appliquent au pilote SAWP CTCG.
Les comités d’éthique sont des organismes indépendants ; leur implication dans le dispositif est donc complexe, mais lorsque cela est nécessaire, le pilote permet aux experts de les solliciter au cas par cas.
Nombre de produits admis dans le pilote
Environ 10 par an, avec un minimum de départ d’un par mois, étant donné qu’aucune réunion n’a lieu en août.
Frais applicables
Les frais standards d’avis scientifique et les incitations tarifaires s’appliquent. Les frais sont identiques à ceux de la procédure SAWP classique ; aucun coût supplémentaire lié au pilote. La confidentialité est assurée selon les principes standards de l’EMA. Les promoteurs ayant complété le pilote recevront un questionnaire afin de contribuer à l’évaluation du succès du pilote.
Résultat : une lettre de conseil scientifique consolidé
À l’issue du processus, le demandeur reçoit une lettre unique de conseil consolidé, incluant les positions scientifiques intégrées du SAWP et du CTCG ainsi que des commentaires spécifiques des États membres (dans une section séparée).
Informations importantes concernant le conseil :
Le conseil n’est pas juridiquement contraignant.
Toute divergence par rapport à ce conseil devra être justifiée dans les futures soumissions de CTA ou d’AMM.
Le conseil ne constitue pas une pré-évaluation des données.
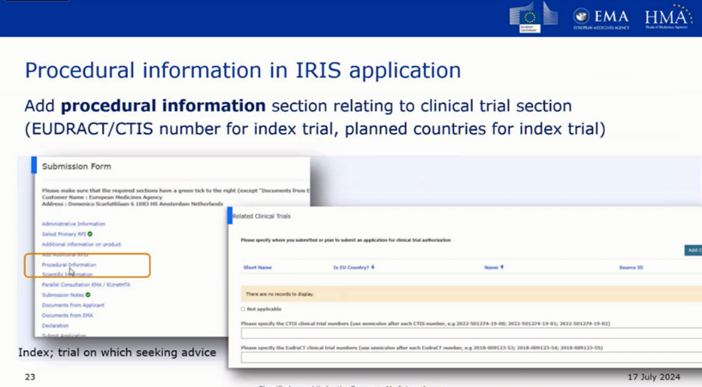
Le pilote Pre-CTA vise à permettre aux promoteurs/demandeurs prévoyant de soumettre une demande d’essai clinique (CTA) via le système d’information sur les essais cliniques (CTIS) d’obtenir des orientations sur les aspects réglementaires et techniques des essais cliniques auprès des évaluateurs des autorités compétentes nationales des États membres de l’UE.
Éligibilité à la procédure Pre-CTA
Critères à satisfaire
La demande doit inclure jusqu’à 5 questions réglementaires/techniques.
Le protocole doit être presque finalisé.
L’État membre rapporteur (RMS) proposé pour la future demande CTIS doit être identifié.
Liste non exhaustive des sujets réglementaires couverts
Le pilote initial comprend cinq procédures, suivies d’une évaluation et d’une optimisation.
Résultat et confidentialité
Le résultat de l’avis est une lettre d’avis non juridiquement contraignante, mais les demandeurs doivent justifier toute divergence par rapport à cet avis lors de la soumission de la demande formelle d’essai clinique.
Il ne s’agit pas d’une pré-évaluation des données.
Les principes de confidentialité et de gestion des conflits d’intérêts s’appliquent.
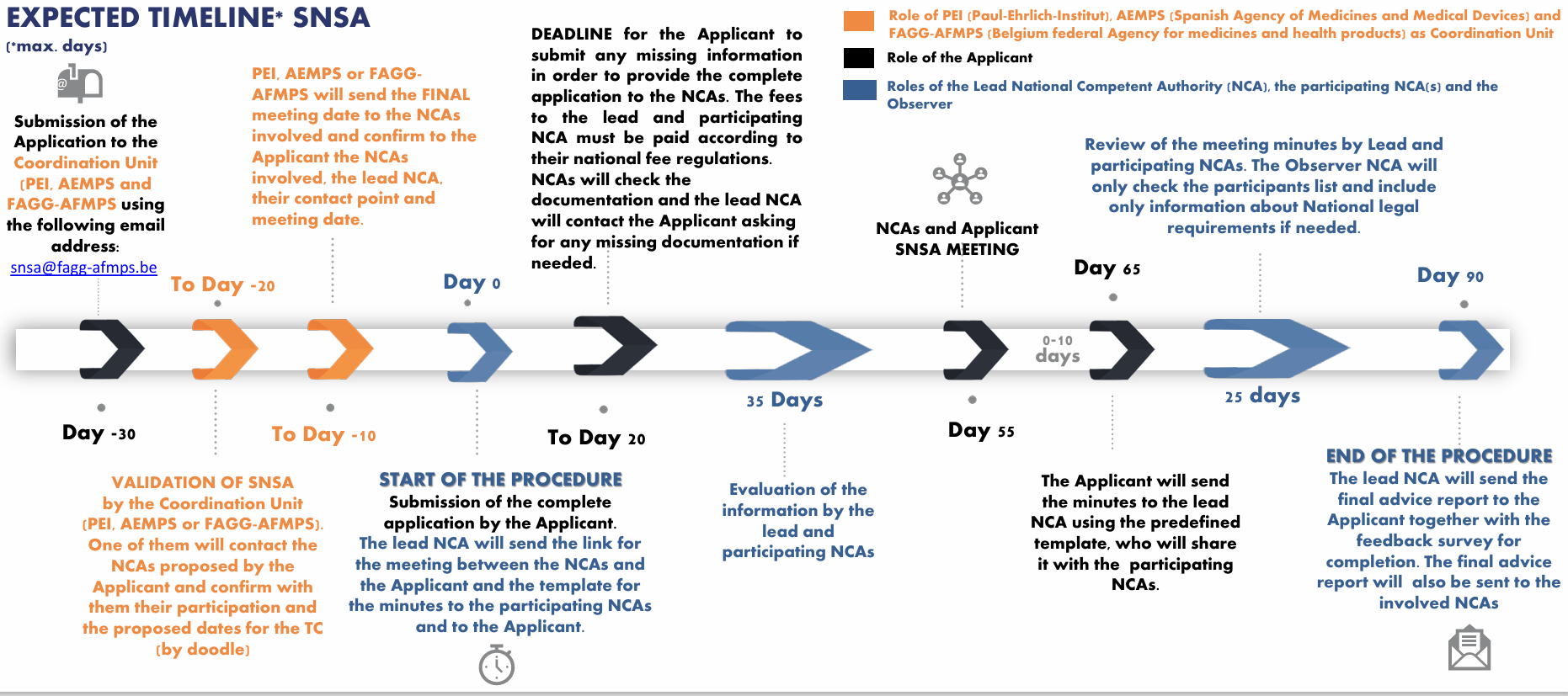
3. Avis scientifique national simultané (SNSA) afin interagir avec plusieurs services d'innovation de différentes agences nationales des médicaments de l'Espace économique européen
Le SNSA est une procédure d’interaction précoce avec plusieurs agences nationales du médicament simultanément. Grâce à cette procédure, un développeur peut obtenir un avis de différentes agences sur la base d’un même ensemble de questions et d’un même dossier de données.
Bénéfices / objectifs
Quels sont les bénéfices pour les agences nationales du médicament impliquées dans le SNSA ?
Comprendre les besoins des demandeurs et stimuler l’innovation.
Faciliter et accélérer l’approbation réglementaire.
Éviter les lacunes dans le soutien réglementaire précoce.
Quels sont les bénéfices pour le développeur ?
Traiter des questions réglementaires scientifiques/techniques spécifiques.
Obtenir des réponses de différentes agences nationales du médicament issues de plusieurs pays en même temps.
Étapes du SNSA
Lors de l’initiation de la demande
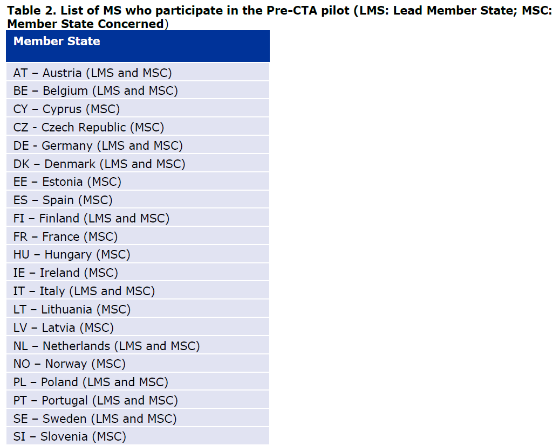
L’un des régulateurs nationaux (au sens des agences nationales du médicament, généralement via leur service d’innovation) doit être choisi comme chef de file parmi la liste des autorités compétentes nationales participantes.
Le chef de file est l’agence responsable de la coordination de la procédure avec les parties impliquées
Un ou plusieurs régulateurs nationaux peuvent participer au SNSA.
Il est désormais possible d’impliquer 3 autorités compétentes nationales (y compris le chef de file), un observateur ou davantage selon les accords en place et si le choix du développeur est justifié.
« Par exemple, lorsque la demande concerne un essai clinique devant être réalisé dans plus de deux États membres, l’implication d’États membres supplémentaires dans une seule procédure SNSA sera envisagée sous réserve de l’accord des autorités compétentes nationales et si cela est dûment justifié par le demandeur. » (Extrait du formulaire de demande publié en mai 2025).
Observateur : l’observateur est autorisé à assister aux réunions, y compris la réunion préparatoire entre les différentes agences nationales du médicament et la réunion formelle avec le demandeur. Toutefois, son rôle ne lui permet pas d’évaluer la demande.
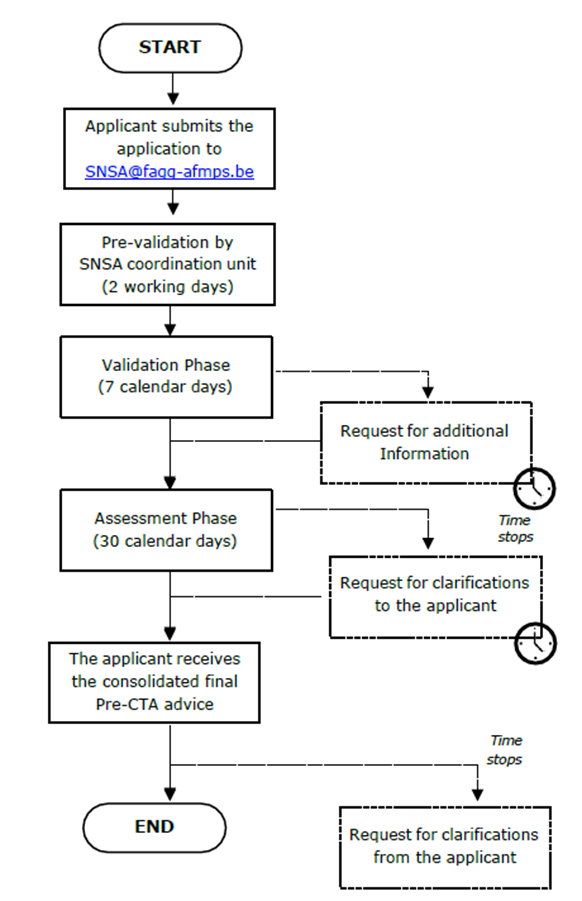
Le développeur doit envoyer la demande de SNSA au point de contact central appelé « unité de coordination » (PEI/AEMPS/FAGG-AFMPS), accompagnée d’un projet de liste de questions issues du formulaire de demande SNSA et d’un bref aperçu du périmètre de la demande.
La liste de questions permet aux régulateurs désignés d’identifier les experts appropriés pour traiter la demande.
Le résumé du périmètre inclut, par exemple, les caractéristiques du produit, le procédé de fabrication (par exemple pour un MTI), le contexte du projet de développement (pré)clinique envisagé, etc.
Début formel de la procédure
Le développeur doit soumettre une lettre de motivation, une liste détaillée de questions accompagnée de la position claire du demandeur pour chacune d’elles, le document de briefing contenant toutes les informations scientifiques et réglementaires détaillées du dossier, ainsi que tout document justificatif supplémentaire.
La procédure commence à l’envoi des documents de briefing.
Jusqu’au jour 20, des mises à jour mineures peuvent être apportées au dossier si elles n’ont pas d’impact sur le choix des experts désignés.
Validation du document de briefing
Il est possible que des demandes d’informations complémentaires, de clarifications ou de re-soumission soient formulées.
La procédure débutera formellement après la validation du document de briefing.
Après validation, la réunion SNSA aura lieu au plus tard dans les 55 jours suivants.
La réunion SNSA
Elle se tiendra dans la plupart des cas de manière virtuelle.
L’agence chef de file animera la réunion.
Aucune nouvelle question ni donnée ne peut être ajoutée pendant la procédure SNSA ou lors de la réunion.
Après la réunion
L’autorité chef de file demandera au demandeur de rédiger le compte rendu de la réunion SNSA formelle en utilisant le modèle fourni.
Le compte rendu devra être transmis à l’autorité chef de file et aux autorités compétentes nationales participantes dans un délai de 10 jours après la réunion.
L’avis consolidé sera transmis au demandeur au plus tard au jour 90 de la procédure.
Dans un délai de quatre semaines, les demandeurs devront fournir un retour sur le pilote SNSA afin d’améliorer le questionnaire et la procédure.
Étapes et calendrier de l’interaction, d’après le site de l’EMA.
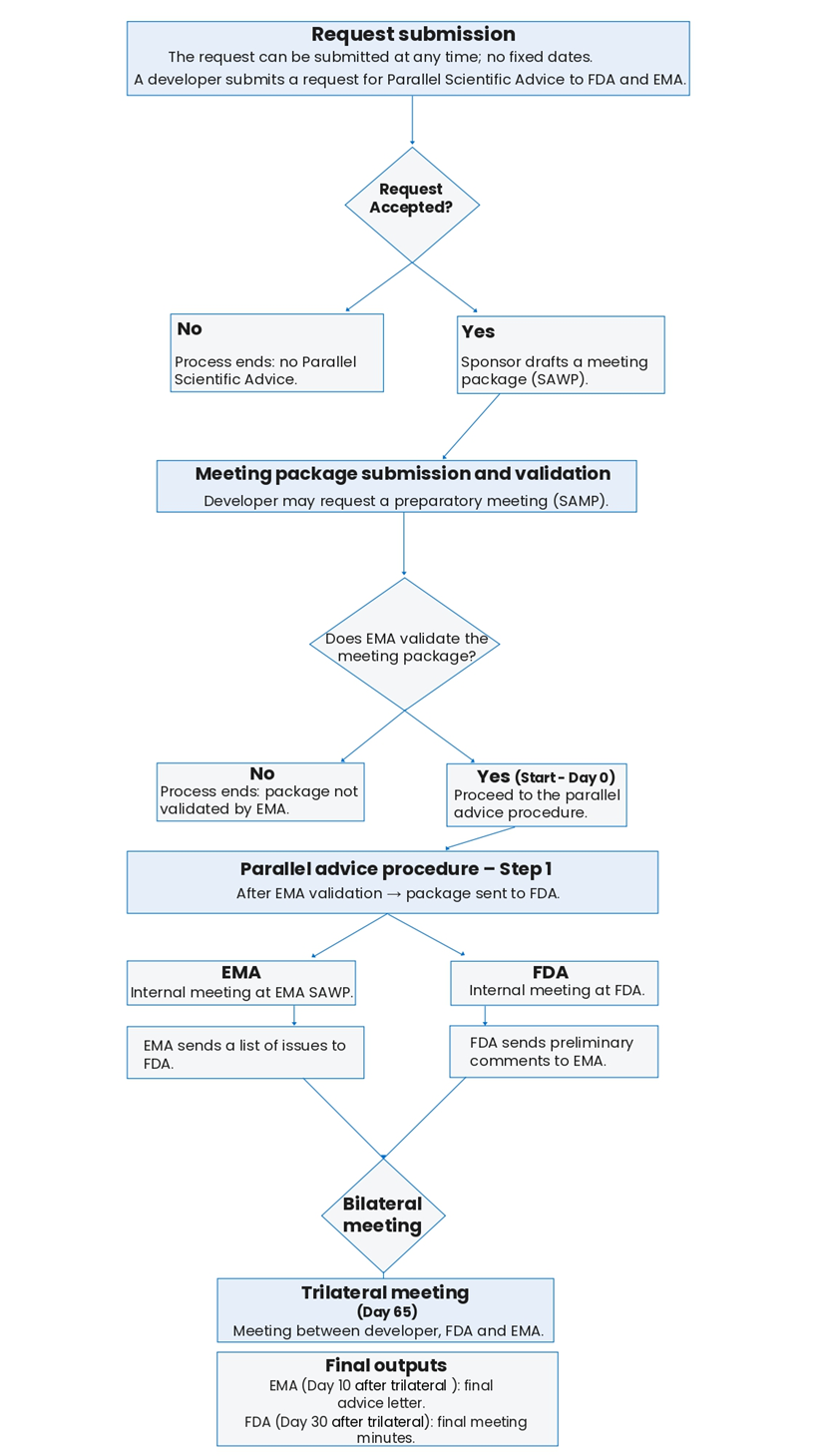
4. Avis scientifique parallèle EMA-FDA pour interagir à la fois avec l’European Medicines Agency (EMA) dans l’Union européenne et la Food and Drug Administration (FDA) aux États-Unis d’Amérique
L’avis scientifique parallèle EMA-FDA vise à obtenir des retours à la fois de l’EMA et de la FDA sur des questions scientifiques et des exigences réglementaires.
Il doit être sollicité au cours de la phase de développement d’un médicament lorsque le développeur cible à la fois le marché de l’Union européenne et celui des États-Unis en vue d’une future autorisation de mise sur le marché et d’une éventuelle commercialisation.
Qui peut demander un conseil scientifique parallèle EMA-FDA ?
La procédure est généralement demandée par un développeur de médicament sur une base volontaire. Toutefois, l’EMA et la FDA peuvent également initier le processus de conseil scientifique parallèle EMA-FDA en pleine coopération avec le développeur dans des circonstances exceptionnelles.
Le développeur de médicament peut être :
le « sponsor » d’une demande d’Investigational New Drug (IND) aux États-Unis,
le « demandeur » soumettant une New Drug Application (NDA) ou une Biologics License Application (BLA) aux États-Unis,
ou
un futur demandeur d’autorisation de mise sur le marché dans le cadre de la procédure européenne.
Étapes vers la procédure
Un développeur peut soumettre une demande de conseil scientifique parallèle à la FDA et à l’EMA à tout moment ; il n’existe pas de dates fixes pour cette demande.
Un développeur soumet une demande de conseil scientifique parallèle à la Food and Drug Administration (FDA) et à l’European Medicines Agency (EMA).
Suite à cette demande, les agences peuvent accepter ou refuser la demande de conseil scientifique parallèle.
En cas de réponse négative, le développeur ne bénéficiera pas du conseil scientifique parallèle et la procédure prend fin.
Si les agences acceptent la demande, le développeur devra préparer un dossier de réunion conformément aux procédures du SAWP.
Soumission du dossier de réunion et phase de validation
À ce stade de la procédure, il est possible pour le développeur de demander une réunion préparatoire conformément aux procédures du SAWP.
Étapes de la procédure de conseil scientifique parallèle EMA-FDA initiée dans l’Union européenne
La procédure commence après la validation du dossier de réunion par l’EMA. Elle dure entre 75 et 95 jours. Toutefois, si l’EMA ne valide pas le dossier, la procédure prend fin.
Après validation par l’EMA, le dossier de réunion est transmis à la FDA. Une réunion interne est organisée à la FDA et une autre au sein du SAWP de l’EMA.
La FDA transmet des commentaires préliminaires sur le dossier à l’EMA, et l’EMA envoie une liste de points à discuter à la FDA.
Une réunion bilatérale est organisée entre les deux agences.
Ensuite, une réunion trilatérale entre le développeur, la FDA et l’EMA est organisée, au cours de laquelle le dossier de réunion et les différentes questions sont examinés et discutés.
Dix jours après la réunion trilatérale, l’EMA adopte la lettre de conseil final.
Trente jours après la réunion trilatérale, la FDA adopte le compte rendu final de la réunion.
Veuillez trouver ci-dessous un organigramme de la procédure d’avis scientifique parallèle EMA-FDA élaboré à partir du calendrier de cette procédure disponible ici.
Figure : Organigramme de la procédure de conseil scientifique parallèle EMA-FDA
Pour plus d’informations sur le calendrier de la procédure, veuillez consulter le document fourni par EMA.
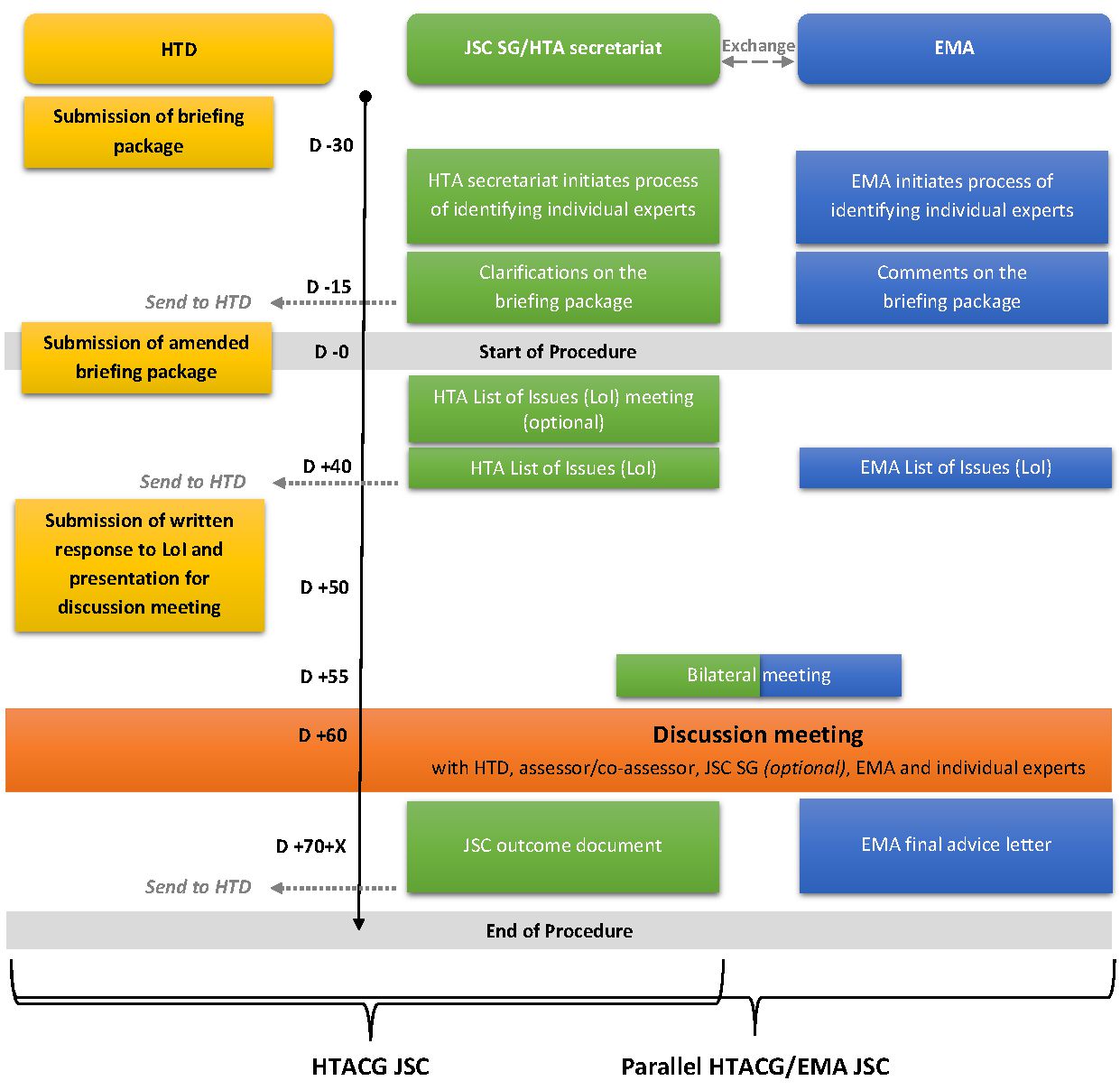
Consultations scientifiques communes (JSC) et consultations scientifiques communes parallèles HTACG/EMA
Articles applicables relatifs aux consultations scientifiques communes du HTACG et aux consultations scientifiques communes parallèles HTACG/EMA : articles 16 à 21 du règlement (UE) 2021/2282du Parlement européen et du Conseil du 15 décembre 2021 relatif à l’évaluation des technologies de santé et modifiant la directive 2011/24/UE (règlement HTA).
5. Consultations scientifiques communes (JSC) pour interagir avec l’ensemble des organismes d’évaluation des technologies de santé (ETS) de l’Union européenne via le groupe de coordination HTA
Les JSC sont coordonnées par le groupe de coordination des technologies de santé (HTACG, Health Technology Assessment Coordination Group). Le sous-groupe nomme des évaluateurs et des co-évaluateurs de différents pays pour superviser le processus. La demande peut concerner un nouveau médicament ou un médicament existant pour une nouvelle indication.
Nombre de réunions par an
Le HTACG tient deux réunions trimestrielles au premier semestre et peut passer à des réunions mensuelles au second semestre (sauf en août). Cette éventuelle augmentation dépend du nombre de comités de pilotage conjoints (JSC) mis en place.
Délai de demande pour JSC
Les dates limites de dépôt des demandes et les instructions relatives à la procédure sont publiées sur le site de la Commission européenne ainsi que sur la plateforme HTA IT
En 2026, les dates limites sont les suivantes :
Du 7 janvier au 4 février 2026
Du 1er au 29 avril 2026
Du 3 juin au 1er juillet 2026
Du 23 septembre au 21 octobre 2026.
Objectif des Consultations Scientifiques Communes (JSCs)
Pour les développeurs des technologies de santé, cette procédure permet de discuter du plan de développement clinique d'une technologie de la santé (ici, un médicament de thérapie innovante) dès les premières étapes. Elle les aidera à produire des données probantes solides.
« Le groupe de coordination organise des consultations scientifiques communes afin d’échanger des informations avec les développeurs de technologies de la santé sur leurs plans de développement en ce qui concerne une technologie de la santé donnée. Ces consultations facilitent la production de données probantes répondant aux exigences en la matière qui sont susceptibles d’apparaître dans le cadre d’une évaluation clinique commune ultérieure concernant cette technologie de la santé. […] Les consultations scientifiques communes concernent en particulier tous les aspects pertinents de la conception de l’étude clinique, ou tous les aspects pertinents de la conception de l’investigation clinique, y compris les comparateurs, les interventions, les résultats de santé et les populations de patients » (Article 16(1)Regulation HTA).
Critères d’éligibilité et critères de sélection pour les JSC applicables aux médicaments
L’éligibilité d’un produit repose sur deux conditions cumulatives :
Les études cliniques et investigations cliniques sont encore au stade de planification.
Le développeur de technologie de santé doit également justifier que le produit remplit un ou plusieurs des critères suivants pour être sélectionné dans une JSC :
Besoin médical non satisfait,
Premier de sa classe,
Impact potentiel sur les patients, la santé publique ou les systèmes de santé,
Dimension transfrontalière significative,
Valeur ajoutée majeure à l’échelle de l’Union,
Priorités de recherche clinique de l’Union.
La réponse concernant l’éligibilité du produit doit être communiquée au développeur dans un délai de 15 jours ouvrables après la fin de chaque période de soumission.
La procédure dure environ 4,5 mois à compter de la réception du document de briefing avec ses annexes et références (briefing package) par le secrétariat ETS via la plateforme informatique ETS. Au jour 60, une réunion de discussion a lieu entre le HTACG et le développeur de technologie de santé.
6. Consultations scientifiques communes parallèles pour interagir avec le groupe de coordination ETS et avec l’European Medicines Agency (consultations scientifiques communes parallèles HTACG/EMA)
Les développeurs peuvent demander à entrer dans une procédure de consultations scientifiques communes parallèles HTACG/EMA lors de leur demande de consultation scientifique commune. Ce mécanisme combine une consultation scientifique commune du groupe de coordination ETS avec un avis scientifique de l’EMA. Cette procédure est établie par l’article 17 du règlement ETS.
Dans cette procédure, les processus du HTACG et de l’EMA se déroulent en parallèle. Bien qu’ils fonctionnent séparément, ils coordonnent les étapes clés de la procédure et partagent les informations nécessaires afin d’assurer le bon déroulement du dispositif. Le calendrier des JSC est synchronisé avec celui de la procédure d’avis scientifique (SA). Les questions posées par le développeur dans cette procédure doivent être différentes de celles ayant déjà fait l’objet d’un avis scientifique récent de l’EMA pour un produit similaire.
Résultat du dispositif
Le résultat consiste en une lettre d’avis scientifique de l’EMA et un document final de JSC du HTACG.
Consultations scientifiques communes au sein du HTACG et consultations scientifiques communes parallèles HTACG/EMA
Règlement (UE) 2021/2282 du Parlement européen et du Conseil du 15 décembre 2021 concernant l’évaluation des technologies de la santé et modifiant la directive 2011/24/UE (texte présentant de l’intérêt pour l’EEE) PE/80/2021/INIT numéro CELEX : 32021R2282
Règlement d’exécution (UE) 2024/3169 de la Commission du 18 décembre 2024 établissant les règles d’application du règlement (UE) 2021/2282 du Parlement européen et du Conseil en ce qui concerne les procédures applicables aux consultations scientifiques communes relatives aux médicaments à usage humain au niveau de l’Union, C/2024/8850 numéro CELEX : 32024R3169
Règlement d’exécution (UE) 2024/2699 de la Commission du 18 octobre 2024 établissant, en application du règlement (UE) 2021/2282 du Parlement européen et du Conseil, les règles de procédure détaillées applicables à la coopération du groupe de coordination des États membres sur l’évaluation des technologies de la santé et la Commission avec l’Agence européenne des médicaments sous forme d’échange d’informations en ce qui concerne l’évaluation clinique commune des médicaments, des dispositifs médicaux et des dispositifs médicaux de diagnostic in vitro et en ce qui concerne la consultation scientifique commune sur les médicaments et les dispositifs médicaux, C/2024/7201, document 32024R2699, numéro CELEX : 32024R2699
Annual Work Programme 2025, adopté le 28 novembre 2024 par le HTACG conformément à l’article 6 du règlement (UE) 2021/2282, V1.0, 28 novembre 2024, HTACG
EMA ACT-EU (Accelerate Clinical Trials in EU),ACT-EU (Accelerate Clinical Trials in EU), conférence ERICA ERN Research, Kristina Larsson, responsable des médicaments orphelins, EMA
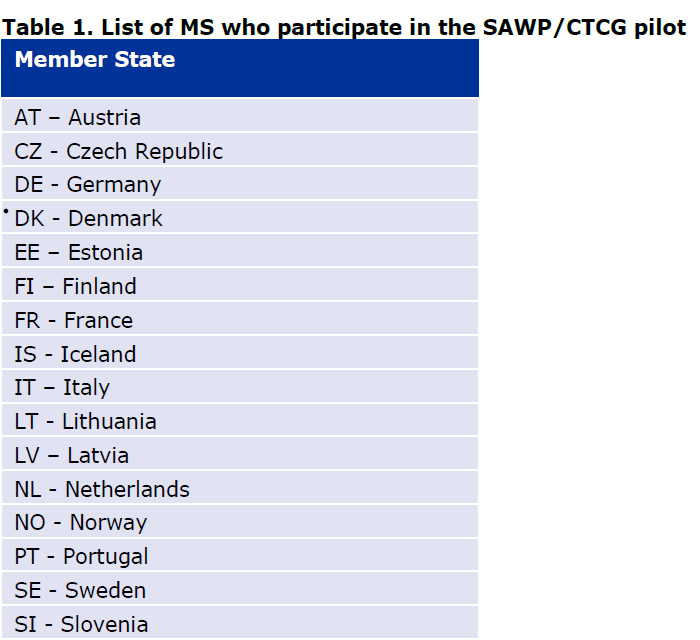
États membres participant aux pilotes ACT EU sur les avis consolidés, PDF, Commission européenne (CE), HMA, EMA