Support from several competent authorities for the development of innovative medicines
Introduction
Early interaction procedures with Competent authorities are implemented to support medicines’ development, mainly innovative medicines, and the specific challenges they face. The support comes in the form of services and regulatory procedures allowing medicines developers to interact with the competent authorities through existing mechanisms during the development of a medicinal product. Early contact with Competent authorities is paramount for developers when working towards making a new medicine to enter the European Union market. With this rationale in mind, multiple services and procedures have been established by Competent authorities to facilitate dialogue or information flow on regulatory questions from developers and to bridge the multiple existing legal frameworks.
On one hand, early contact can be established with only one competent authority, either at the European Union level or at the national levels, using these procedures or schemes. On the other hand, some of these schemes are designed to allow developers to enter into contact early on with several Competent authorities together at the same time. When they involve multiple competent authorities, the interactions can be multi-level when occurring within the European Union. For instance, with the European Medicines Agency (EMA) and one or several National Competent Authorities (such as Health Technology Assessment (HTA) bodies in the European Union or National Medicines Agencies). These procedures can also target a specific step of the research and development timeline, such as clinical trials for example (Pre-CTA or SAWP-CTCG advice pilots). They can also target two authorities in different regions of the world, here EMA for the European Union market and the Food and Drug Administration (FDA) for the USA market. Early interactions can also take place with multiple competent authorities on a National scale (such as for Simultaneous National Scientific Advice).
Six different procedures can be considered as procedures with multiple competent authorities. They will be introduced here as such:
Scientific Advice from the Scientific Advice Working Party (SAWP) Clinical Trials Coordination Group (CTCG) Pilot to interact with all participating National medicines agencies through EMA on the suitability of a clinical trial design to support a Clinical Trial Application (CTA) within a Member State or to support developers with their Marketing Authorisation Application (MAA);
Pre-CTA Pilot is coordinated by CTCG to interact with all participating National medicines agencies on regulatory issues on the Clinical Trial Application (CTA) dossier before submitting a Clinical Trials Application on CTIS;
Simultaneous National scientific advice (SNSA) to interact with two or more innovation offices from National medicines agencies of the European Economic Area (EEA);
Parallel scientific advice to interact with both the EMA and the FDA.
Joint Scientific Consultations to interact with all Health Technology Assessment (HTA) bodies from the European Union via the HTA Coordination Group;
Parallel Joint Scientific Consultations to interact with several HTA bodies from the European Union and with the EMA;
Main Actors
ACT-EU: ACT-EU was created by a joint action from the European Medicines Agency (EMA), the Heads of Medicines Agency (HMA) and the European Commission (DG SANTE and DG RTD). It was established in January 2022 with the impulse of the implementation of the new clinical trials regulation (CTR). ACT-EU has a workplan with 11 priority areas. Find out more about ACT-EU on their website. You can find the workplan for 2023-2026 and the latest yearly workplan for 2025-2026. The ACT EU workplan 2025 – 2026 explains clearly the aim of this action is to accelerate Clinical Trials in the European Union by supporting smarter clinical trials using multiple processes (innovation, regulatory and technological) to foster innovation. “The vision is to have better, faster and optimised clinical trials, benefitting patients and healthcare in Europe. Seamless coordination between stakeholders, regulators and ethics committees strengthens cross-border collaboration. The ACT EU multi-stakeholder platform is central to this”.
Clinical trials coordination group (CTCG): The CTCG is a working group at the Heads of Medicines Agencies (HMA). It is composed of experts from National Medicines Agencies on the assessment and oversight of clinical trials. This group aims at enhancing the attractiveness of EU/EUA for clinical trials by promoting harmonization and optimization of regulations. Find out more about this group here. To read the CTCG mandate follow this link.
EU innovation network (EU-IN): A working group created in 2015 to support the European Medicines and Regulatory Network (EMRN) on the development of new innovative medicines. It works under the joint mandate of the EMA and HMA, and includes representatives from national innovation offices. It aims to facilitate the development of innovative products at national and European levels, including to favour the use of the existing regulatory support schemes, such as scientific advice. It was designed to strengthen the collaboration between National Medicines Agencies and the EMA on regulatory matters relating to emerging therapies and associated technologies. (from EU Innovation Network (EU-IN) | European Medicines Agency (EMA)). This group has also horizon scanning activities to find and define the new emerging trends in the field of medicines. A dedicated subgroup is in charge of the Simultaneous National Scientific Advice scheme led by the Paul Ehrlich Institute (PEI), the National Medicines Agency in Germany. This subgroup has been running SNSA procedures since 2020. The EU-IN is also in charge of promoting competent authorities' involvement in externally funded projects. (See, Considerations for research / project teams seeking competent authority participation in externally funded regulatory science and public health research projects related to medicinal products, 2023 EMA/38599/2023, 19 January).
European Medicines Agency: The European Medicines Agency (EMA) is an agency of the European Union whose goal is to protect and promote human and animal health. The agency is responsible for the scientific assessment of Advanced Therapy Medicinal Products to be marketed within the European Union and the European Economic Area. The Agency provides a number of incentives and procedures for medicine developers to support better patients’ access to medicines.
European Medicines Regulatory Network (EMRN): The European Medicines Regulatory Network is a coordinated regional regulatory system where the European Commission, the European Medicines Agency and the Medicines regulatory authorities from the 30 European Economic Area (EAA) countries work together. The network ensures that the medicines authorised are safe, effective and high-quality medicines. More information can be found on EMA’s website under 'European medicines regulatory network'.
Heads of Medicines Agencies (HMA) : The Heads of Medicines Agencies is a network composed of the heads of the National Medicines Agencies. National Medicines Agencies are responsible in their own country for the regulation of medicines for human and veterinary use in the European Economic Area. The HMA is working in cooperation with the EMA and the European Commission to strengthen European cooperation on regulatory activities. For more information on the institution or its tasks follow this link.
Health Technology Assessment (HTA) bodies : Organisms in charge of evaluating health technologies (such as a medicine, a medical device, a medical or surgical procedure). The provided assessment includes the summarized information on medical, patient, social, economic and ethical aspects, and on the consequences of the use of a health technology. (See the definitions of Health Technology Assessment and Health technology in EuroGCT’s glossary of terms)
HTA Coordination Group (HTACG): A group composed of EU Member States’ representatives, (mainly from HTA authorities and bodies) and established by the EU Regulation on Health Technology Assessment. Its key tasks are to coordinate and adopt the joint HTA work carried out by its sub-groups within the scope of this Regulation and to adopt methodological and procedural guidance documents for joint work. The HTACG also aims to ensure cooperation between the relevant European Union bodies (e.g. the European Medicines Agency and HTA bodies), as well as appropriate involvement of stakeholder organisations and experts in its work. (Find out more about the HTA coordination group). The HTA coordination group is notably responsible for the Joint Scientific Consultations presented below.
Innovation offices: Innovation offices are a service provided by National medicines Agencies. They centralise the services and procedures available to developers of innovative health products (such as innovative medicines), including the ones designed to facilitate early interactions with regulators. Their aim is to help developers navigate the different support schemes that are available, and choose the right one according to their specific need. Innovation offices are a way of targeting and answering developers needs and to give them the support they need to bring new innovative medicines to the market. Examples of schemes and procedures for which innovation offices provide guidance are: the scientific advice procedure at the national level, or the simultaneous national scientific advice (SNSA) procedure where available. It is important to underline that not all countries in the European Economic Area (EEA) have such a designated entity to assist developers in obtaining the information they need when creating a new medicine. The list of existing Innovation offices in the European Economic Area (EEA) countries is available on the EuroGCT Actors and Networks database.
Medicine developers: Any entity or legal person aiming to bring medicines to patients. Medicine developers are the target of early interaction with regulators such as National medicines agencies or the European Medicines Agency (EMA), but also HTA bodies. Medicine developers can be SMEs, academia, big pharma, or non-profit organisations.
National medicines Agencies : At the national level, National medicines Agencies are the regulator in charge of promoting human health and ensuring the safety of medicines. They are the public body responsible for regulating medicines under the authority of the ministry of health in most countries. National medicines agencies evaluate, authorise, regulate and control human medicines as well as health in the realm of the State. A comprehensive list of National Competent Authorities in EU Member States can be found in EuroGCT’s Actors and Networks database.
Regulators: Entities designated by law for authorisation, approval, evaluation, oversight, surveillance, and/or adoption of guidance in a specific field. In the context of gene and cell therapies, multiple regulators are involved throughout the development cycle: medicines agencies (European or National), Health Technology Assessment bodies, research funding and authorisation authorities, patent offices, ethics bodies/committees, notified bodies (in the context of combination with medical devices), data authorities, etc. Please note that in most of EMA documents, the term ‘regulators’ is used to refer to the EMA and National medicines agencies only. (From EuroGCT’s Glossary of terms)
Scientific Advice Working Party (SAWP): An EMA working group set up by the Committee for Medicinal Products for Human Use (CHMP) to provide scientific advice and protocol assistance to developers. It is one of the major actors for early interaction between the EMA and stakeholders. The SAWP is involved in the procedure of Parallel scientific advice with EMA and FDA. It is also involved in the SAWP-CTCG pilot procedure created by the ACT-EU initiative. For each request for scientific advice or protocol assistance two delegates are appointed as coordinators of the assessment, and at least one as peer-reviewer of the final advice (See Human Medicines Division Mandate, objectives and rules of procedure of the Scientific Advice Working Party (SAWP) 14 April 2025 EMEA/CHMP/SAWP/69686/04 Rev 19). More on SAWP on the EMA website here.
Seeker of interaction: The medicine developer interested in a specific procedure or service established to foster early interactions between Competent authorities and stakeholders. The purpose of the interaction is to facilitate the development of future marketable medicines.
Food and Drug Administration (FDA): The Food and Drug Administration is the National Medicines Agency for the United States of America. It is responsible for protecting and promoting public health, notably through the assessment and approval of medicines and medical devices to be marketed in the United States.
Definitions
Adaptive Pathways: The ‘Adaptive pathways’ was part of the EMA strategy to encourage prompt access to medicines for patients in need. This regulatory procedure was built for a progressive and adapted patients’ access to medicines. It used other already existing regulatory procedures, such as scientific advice. The pilot project for the adaptive pathways ran from 2014 to 2016. It gave a platform for developers to have informal discussions with the EMA and other stakeholders, patients and HTA bodies for instance to reflect on scientific and regulatory questions. The procedures helped the developers to ask questions and directed them towards another form of early interaction if needed, scientific advice for instance. It was replaced by another procedure for early interaction: parallel scientific advice with HTA bodies which has now been morphed into Parallel joint scientific consultations following the adoption of the HTA Regulation in 2021. For more information on adaptive pathways.
Early interaction: Possible contact between medicines’ developers and Competent authorities through dedicated procedures or services at an early stage in the development chain of a medicine (mostly at the research and development phase). The procedures or services vary depending on the type of product, the statute of the medicines’ developer, and the needs at stake. Early interactions are generally voluntary schemes developers can choose to enter in, none are compulsory.
Innovative medicine: A medicine that contains an active substance or combination of active substances that has not been authorised before (from EMA Glossary of Regulatory terms here.)
Joint Scientific Consultation (JSC): The voluntary procedure allowing a medicine developer to obtain individual guidance on the information, data, analyses and other evidence that are likely to be required from clinical studies by National competent authorities that will assess and/or decide on medicines’ reimbursement by the National health insurance system. This procedure is conducted at the European level via the HTA Coordination group composed of Member States’ representatives from HTA authorities and bodies. It might facilitate the future mandatory procedure of joint clinical assessment of medicines authorised at the EU level, the result of which should be given due consideration by National competent authorities when assessing and deciding on medicines’ reimbursement.
Parallel Joint Scientific Consultation with EMA and health technology assessment (HTA) bodies: A voluntary procedure allowing medicine developers to obtain feedback from both the EMA and HTA bodies, via the HTA coordination group at the EU level, on their plans for evidence generation regarding both marketing authorisation and the reimbursement of medicines.This procedure focuses on overcoming the challenges raised from different regulated activities (marketing authorisation and reimbursement of medicines) with decision making at different levels, at the European Union level for the decision on marketing authorisation of ATMPs and National levels for the decisions on reimbursement of ATMPs. This procedure helps developers on their evidence-based plans regarding applications for marketing authorisation and reimbursement of new medicines. It helps developers to generate solid evidence simultaneously for different Competent authorities regarding different regulated activities, which are parts of the chain to bring ATMPs to the patients. Indeed, the EMA is in charge of the scientific assessment of ATMPs for marketing authorisation, and the HTA bodies, represented in the HTA coordination group at the EU level, are in charge of the assessment of ATMPs for reimbursement decisions. The procedure of Parallel Joint Scientific Consultation builds on previous pilot initiatives and collaborations between Medicines agencies and HTA bodies, led by EMA, EUnetHTA and the European Commission. They include the Report of the pilot on parallel regulatory-health technology assessment scientific advice, EunetHTA's early dialogue initiative, the SEED project (2008-2013) funded by the European Union and joint research on levels of alignment between medicines regulators and HTA bodies in parallel advice. For more information see: Partners and networks: Health technology assessment bodies.
Parallel Scientific Advice (EMA-FDA Parallel Scientific Advice): An optional and voluntary procedure for medicine developers to benefit from the advice of both the European Medicines Agency in the European Union and the Food and Drug Administration in the USA. Generally used during the research and development phase of the product’s life cycle. EMA-FDA Parallel Scientific Advice allows developers of complex products to gain precious time when considering to apply for marketing authorisations in both the United States and the European Union. Indeed, they can benefit from the advice of multiple competent authorities, in different regions, meaning with different regulations to follow and apply.
Scientific Advice from Scientific Advice Working Party (SAWP)- Clinical Trial Coordination Group (CTCG): Rationale behind the creation of the consolidated pilots on clinical trials including the SAWP-CTCG pilot is to provide a procedure to help create better, faster, optimized clinical trials in the EU. This pilot is meant to provide and enhance advice on scientific aspects of clinical trials and development program within the European Medicines Regulatory Network (EMRN). It is supposed to help developers with the Clinical trial and the Marketing Authorisation applications. This is possible through the collaboration between the European Medicine Agency’s (EMA) SAWP and the CTCG involving the Member States and their National Medicines Agencies.
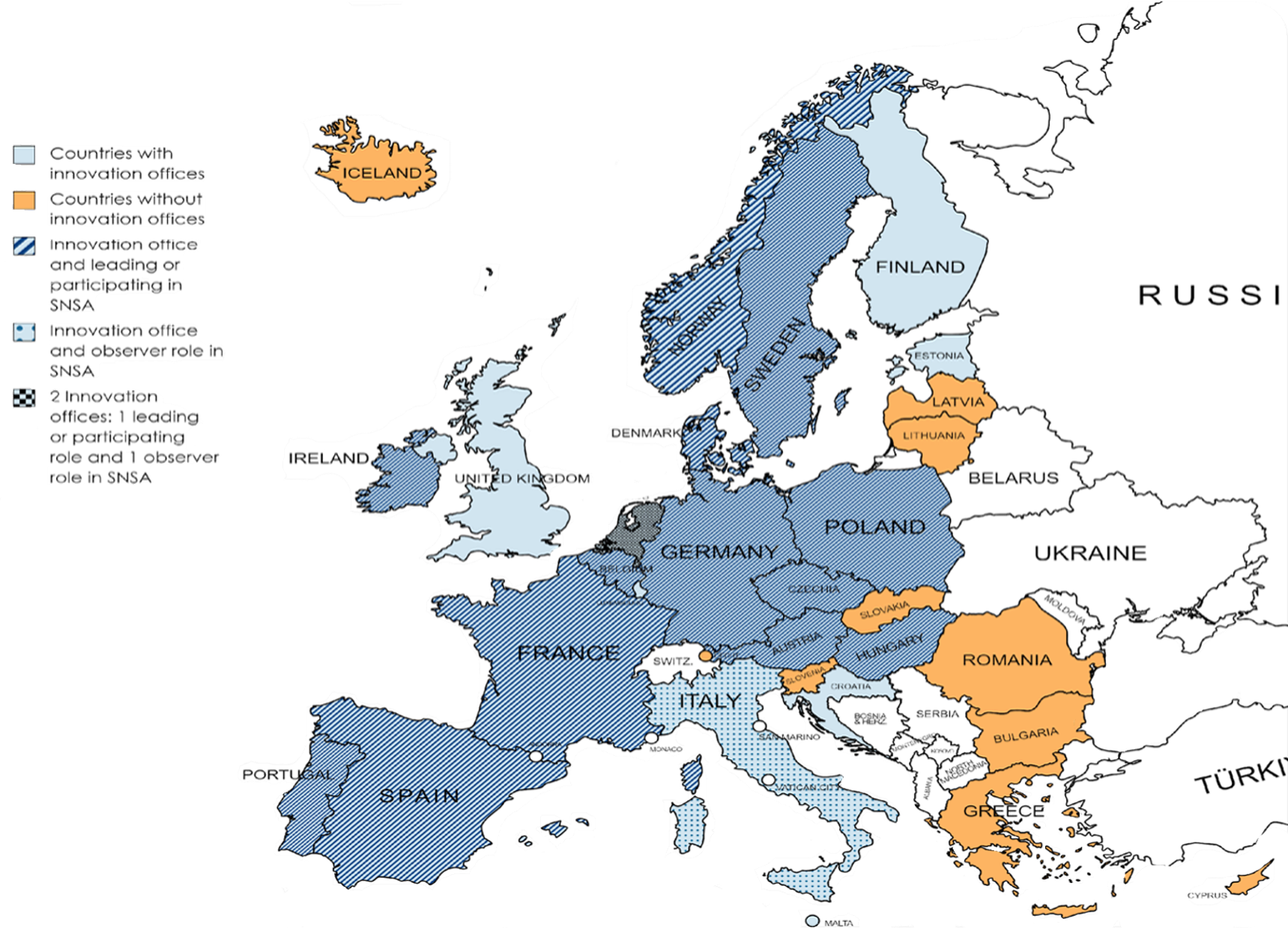
Simultaneous National Scientific Advice (SNSA): A procedure led by the Heads of Medicines Agencies (HMA) and the European Medicines Agency (EMA) for developers wishing to get scientific advice from multiple National competent authorities at the same time. It started as a pilot procedure designed to be time and cost efficient for developers in February 2020. It entered an optimisation phase in 2025. It allows developers to enter into contact and ask for national scientific advice or regulatory advice with two (or more if the need justifies it) National competent authorities (through their innovation office) from different European countries at the same time as long as they are enrolled in the scheme. This is a complementary tool to existing possibilities of scientific advice at the EMA only, and regulatory advices at the national level in each country. Participating countries are mapped below as well as the innovation offices in EEA countries. This map shows how innovation offices are often linked to participation in SNSA, even though some countries with an innovation office are not participating in SNSA (e.g. Estonia, Finland, Croatia and the United Kingdom). However, all countries participating in SNSA have an innovation office showing a clear need for this service to implement the SNSA procedure.
Source: A. DELAGE, L.-S. GILBERT, A. MAHALATCHIMY, Mapping regulators’ early interactions procedures to support innovation, European Society for Gene & Cell Therapy Congress 2023, Brussels, Belgium, 24-27 October 2023, poster.
STARS: the EU-funded Coordination and Support Action (CSA) on the Strengthening Training of Academia in Regulatory Science (STARS) that ran from 1st January 2019 to 30th June 2022. It was meant to support the development of innovative medicines from Academia in giving them out the necessary regulatory support. It aimed to reach out to medicine innovators in academia, bridge the regulatory knowledge gap and enhance dialogue between academia and regulatory authorities. This document available on the EMA website shows the scope of the action and the partners involved. Find more information on the action on the STARS website.
Challenges
Coordinating the different requisites from various regulators and knowing whom to ask the questions regarding to their competency can be an obstacle to the development of a medicine but schemes involving multiple regulators can help to overcome this issue.
For developers trying to design a new medicine, knowing, understanding and complying with the many regulatory requirements at the national level and at the EU level is a primary challenge. They need to navigate the different legal and regulatory requirements and understand how to articulate them.
Moreover, developers may want to know how to access different markets on a global scale and the rules can differ as the requirements are different from one market to another. For instance, to access the European Union market and the United States market, the rules are different. Therefore, if a developer wants to access both markets, they may need to enter into early interaction with both of the “Regional” Competent authorities to understand the necessary scientific and regulatory requirements. With this objective in mind, the EMA and the FDA have created a type of joint scientific advice called “Parallel scientific advice” between the two agencies on scientific issues.
Sometimes competent authorities are bridging the developer’s knowledge gap by creating new schemes and procedures to help them bring new and innovative medicines to patients. The different initiatives may evolve rapidly, because of the speed of development of the field or the experience gained. Thus, it could be difficult to know, understand, and follow the evolution of the different procedures available from various competent authorities at various levels when necessary (national, European, and in other regions of the world).
Moreover, the names or explanations of several procedures may be confusing. For a long time, the phrase parallel advice was the name for two types of procedures, the previously mentioned scheme between EMA and FDA and the one between EMA and HTA Bodies. It created unnecessary confusion. Since the entry into force of the new HTA Regulation, the vocabulary used and the process has been clarified. Indeed, the procedure to interact with multiple HTA bodies is now named Joint Scientific Consultations (JSCs). Moreover, it is still possible to combine consultations from both the EMA and HTA bodies at the same time through Parallel joint scientific consultations. In addition, the procedure to interact with both the EMA and FDA is called Parallel scientific advice with the United States or with the FDA. Despite an effort from clarification initiated by the HTA Regulation and expanded by the EMA, the term “Parallel” is still used in two very different procedures although it always implies the involvement of the EMA with another regulator.
Finally, the costs of procedures may be an obstacle, even if some procedures are free of charge, and incentives exists for SME’s and Academia, depending on the competent authorities involved and the procedure targeted.
Opportunities and incentives
Competent authorities or regulators, are creating services and procedures to facilitate answers from multiple regulators to medicine developers. The primary aim is to support medicine development and medicine’s access to patients. An implicit aim is to avoid losing ground-breaking research to the hardships of navigating the regulatory pathways in regards to the specific area of competencies and the specific regulation or legislation applicable.
There is a strong will to make different authorities come together, some countries having a history of collaboration through bilateral agreements and specific procedures.
Some international activities from agencies can allow new programs to emerge and therefore favor new collaborations, and ultimately the development and the access to new medicines. For instance, the FDA is looking into a new pilot program called Gene Therapies Global Pilot (CoGenT) that will initially include regulatory members of the International Council for Harmonisation (ICH), including the EU, the US, Japan, Canada, and Switzerland. This program is a collaboration between the standing regulatory members of the ICH. The partners can participate in internal regulatory meetings or regulatory meetings with the developer asking for the interaction. This collaboration pilot was mentioned in the ‘CY 2024 report’ from the director of the FDA (read the entire report here). The aim of this pilot is to work towards international regulatory convergence. It seems to be just a plan for now but it shows the importance given by multiple countries in working together on regulatory issues and the standardisation of regulations across different markets to help innovative medicines reach the patients.
It is essential that stakeholders are aware of the existence of these procedures and their development. A number of events are organised by regulators to create such awareness among stakeholders. Some are listed through a regular legal news watch on the ELSIBI website. There are a clear interest and a will to enhance support to stakeholders in facilitating their interactions with regulators or competent authorities more widely by creating more spaces for conversations. It pushes competent authorities to create new procedures and clarify the requirements for a medicine to access the market or to have the most chances at accessing the market. For example, the existence of innovation offices in itself is a marker of interest for a lot of countries and of competent authorities to support the development of new innovative medicines. Also, there seems to be a will for common approaches by identifying the regional challenges and creating common procedures. For instance, the SNSA procedure is one example of a possible way to bring closer support systems regarding current issues as similar questions arose and similar evidences are needed from different National competent authorities.
Finally, there are other support systems available. It exists collaborations between several countries in the form of specific agreements such as the BENELUXA agreement between Belgium, Austria, Ireland, Luxembourg and the Netherlands in Europe (Find out more on the Beneluxa website). This partnership aims to ensure sustainable access to innovative medicines at affordable costs by bringing together multiple countries. The idea is to facilitate dialogue and the exchange of information on medicine policies in the hopes that it will have positive repercussions on medicine’s costs and policies. It also allows countries to negotiate prices with the industry together and therefore increases transparency between different countries on the pricing of medicines.
Practical steps
Developers can enter into contact with their National Medicines Agencies but also with the Heads of Medicines Agencies (HMA) and its working groups, HTA bodies, the EMA or/and the FDA depending on their aim and the type of interaction they need to pursue in order to commercialise their product.
Six different procedures currently co-exist and allow developers to interact directly with several regulators:
If the interaction sought concerns clinical trials and the developer seeks scientific advice on both clinical trial application requirements and Marketing authorisation requirements, they should consider asking for advice through the Scientific Advice from SAWP CTCG Pilot.
If the interaction sought concerns a developer looking for regulatory advice on the Clinical Trial Application (CTA) dossier before submitting a Clinical Trials Application on CTIS, they could use the Pre-CTA advice pilot.
If the interaction sought concerns clinical trials to be conducted in more than one EU Member State, the medicine developers (often the sponsor, please see Clinical trials) should consider entering a procedure of Simultaneous National Scientific Advice (SNSA) to interact with several innovation offices from different National medicines agencies in the European Economic Area.
If the interaction sought concerns the evidence generation for the marketing authorisation of an ATMP in both the EU and the USA markets, the medicine developer should consider to start a procedure of EMA-FDA Parallel Scientific Advice.
If the interaction sought concerns the reimbursement of an ATMP (or more widely a health technology) in several EU member States, the developer should consider using the Joint Scientific Consultation procedure to interact with all HTA bodies from the European Union via the HTA Coordination Group.
If the interaction sought concerns the evidence generation for both the marketing authorisation at the EU level and the reimbursement of an ATMP in several EU member States, the medicine developer should consider to start a procedure of Parallel Joint Scientific Consultations to interact with the HTA Coordination group and with the EMA.
Please find below more details on the practical steps for each one of these procedures.
Consolidated advice pilots on Clinical Trials
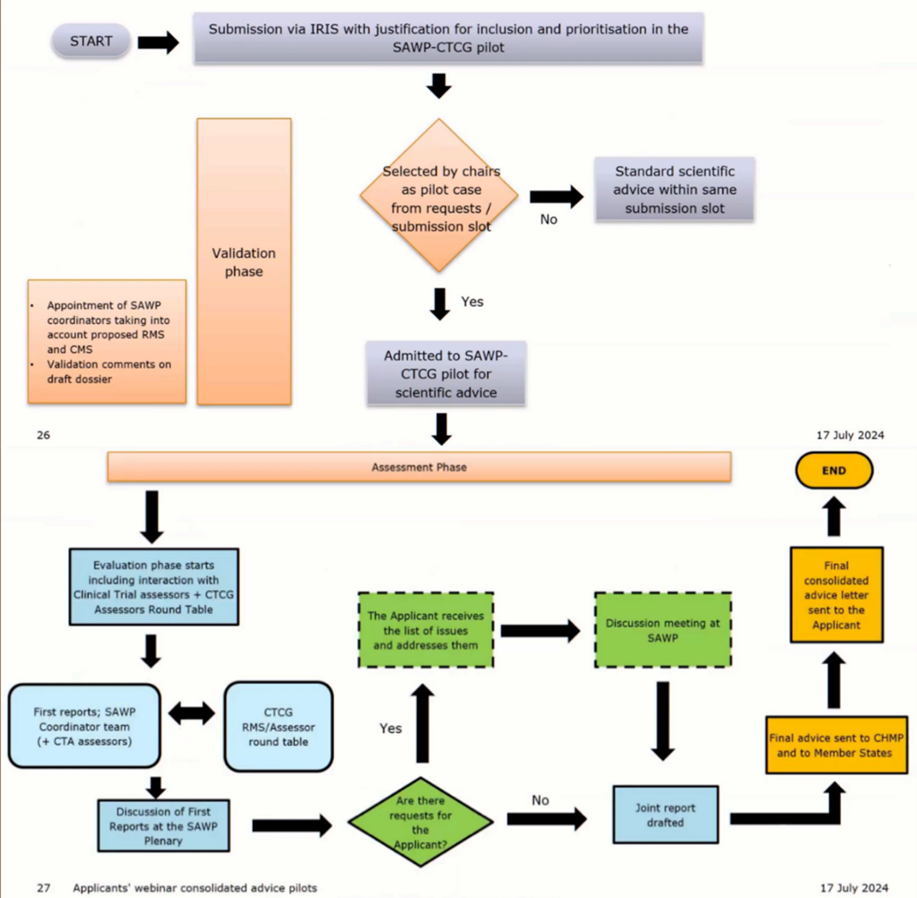
1. Scientific Advice from SAWP CTCG Pilot
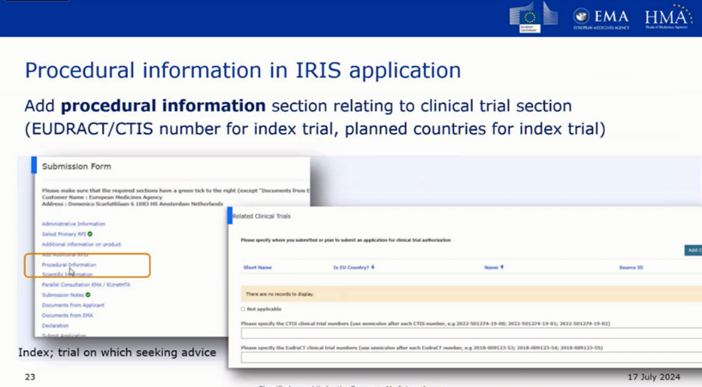
The SAWP CTCG Pilot is a type of Scientific Advice on the suitability of a clinical trial design to support the Marketing Authorisation Application (MAA) at the European level if the pathway is the centralised process and/or clinical trials applications (CTA) to a National medicines Agency within a specific Member State. The pilot aims at creating harmonised view and advice on clinical trials designs for both applications (CTA and MAA) across EU Member States. It is based on an enhanced collaboration between the European Medicine Agency’s (EMA) Scientific Advice Working Party (SAWP) and the Member States’ Clinical Trial Coordination Group (CTCG).
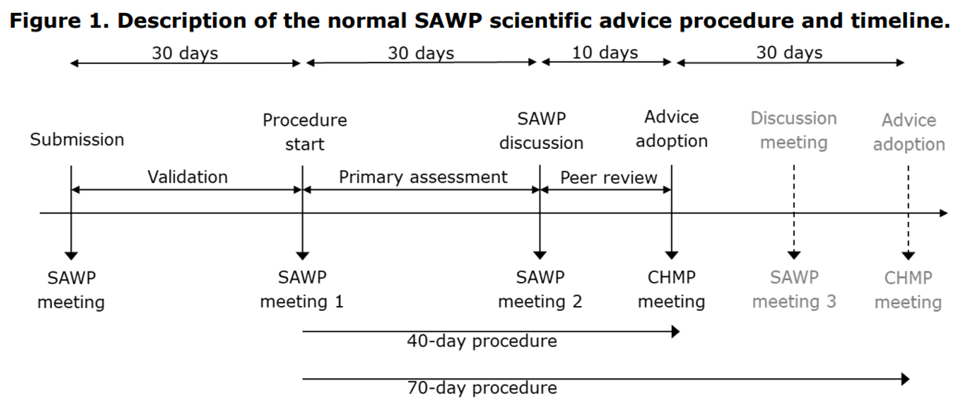
The normal SAWP scientific advice procedure and timeline apply to the Scientific Advice from SAWP CTCG Pilot.
Ethics committees are independent bodies therefore their involvement in the scheme is complex but when needed the pilot allows the experts to reach out on a case-by-case basis.
Number of products admitted to the pilot
Approximatively 10 per year with a minimum starting number of one by month considering there is no meeting in August.
Applicable fees
Normal Scientific advice fees and fees incentives apply. Fees are identical to standard SAWP advice; no additional pilot charges. Confidentiality follows standard EMA principles. Sponsors completing the pilot will receive a questionnaire to support evaluation of pilot success.
At the end of the process, the applicant receives a single consolidated advice letter, including SAWP and CTCG integrated scientific positions and MS‑specific comments (in a separate section).
Some important information on the advice:
The advice is not legally binding.
Any deviation from the advice must be justified in future CTA or MAA submissions.
The advice does not constitute a pre-assessment of data.
The Pre-CTA advice pilot aims for sponsors/applicants planning to submit a Clinical Trial Application (CTA) to the Clinical Trial Information System (CTIS) to obtain guidance on the regulatory and technical aspects of clinical trials from assessors of the National Competent Authorities of EU Member States.
Eligibility to apply for the Pre-CTA advice pilot
Criteria that need to be satisfied
The request should include up to 5 regulatory/technical questions.
The applicant must have an almost mature protocol.
The proposed Reporting Member State (RMS) for the future CT application in CTIS must be known.
A non-exhaustive list of regulatory topics that are covered by the Pre-CTA advice pilot
The Initial pilot includes five procedures, followed by a review evaluation and a following optimization of the process.
Outcome and Confidentiality
The outcome of the advice is a non-legally binding advice letter but applicants should justify why they may have strayed from the given advice when submitting the formal Clinical Trials Application.
It is not a pre-assessment of the data.
The confidentiality and conflict-of-interest principles apply.
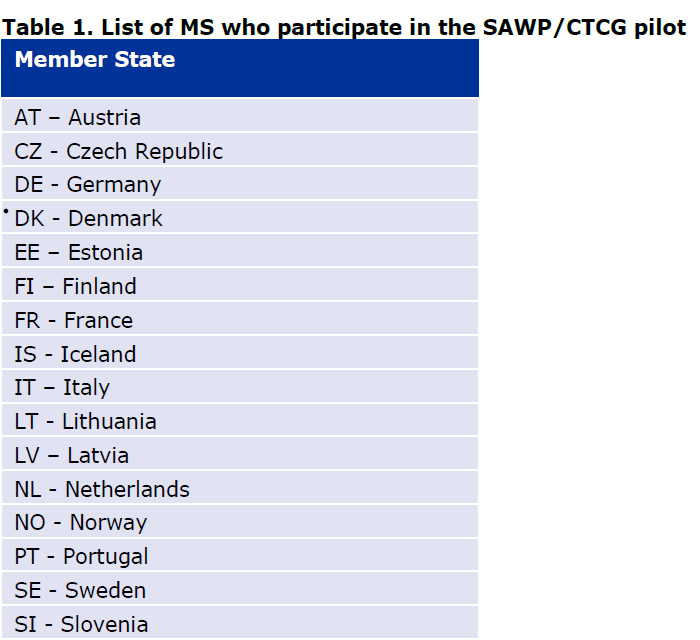
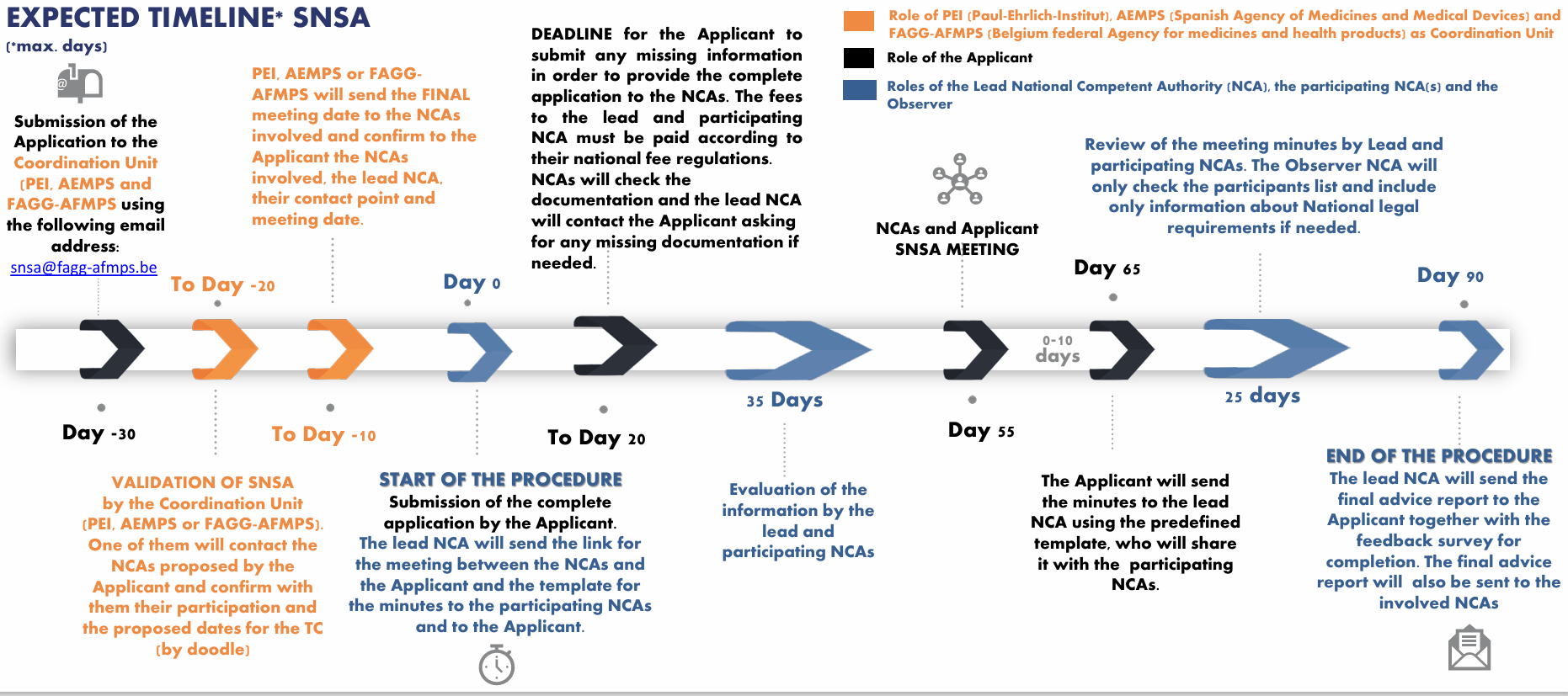
3. Simultaneous National Scientific Advice (SNSA) to interact with several innovation offices from different National medicines agencies of the European Economic Area
SNSA is a procedure for early interaction with multiple National Medicines Agencies, at the same time. With this procedure a developer can obtain an advice from different agencies with the same set of questions and data package.
Benefits/aim
What are the benefits for the National Medicines Agencies involved in SNSA?
To know the needs of the applicants and enhance innovation.
To address specific scientific/technical regulatory issues.
To get answers from different National medicines agencies from multiple countries at the same time.
Steps for SNSA
When initiating the submission request
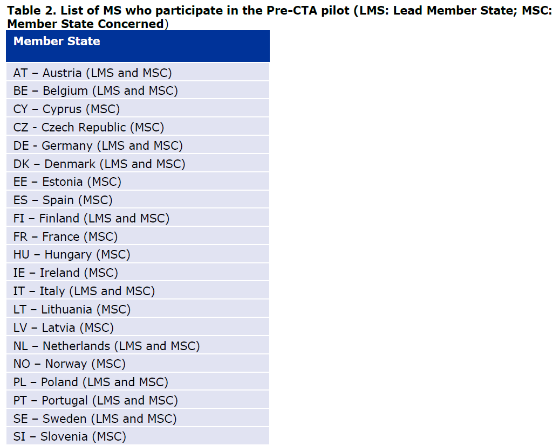
One of the national regulators (here in the sense of National medicines agencies, mostly through their innovation office) should be chosen to be the lead from the list of participating National Competent Authorities.
The lead is the agency responsible for coordinating the procedure with the involved parties.
One or more national regulator can take part in the SNSA.
It is now possible to involve 3 National competent authorities (Including the leader), an observer or more depending on the agreements in place and if the developer’s choice is justified.
"E.g. where the request relates to a clinical trial to be performed in more than 2 MSs, the involvement of additional MSs in a single SNSA procedure will be considered subject to the agreement of the NCAs and if adequately justified by the Applicant.” (Extract from the application form published in May 2025).
Observer: The observer is allowed to attend the meetings including the preparatory meeting between the different National medicines agencies and the formal meeting with the applicant. But the role of observer in the procedure does not allow to evaluate the application.
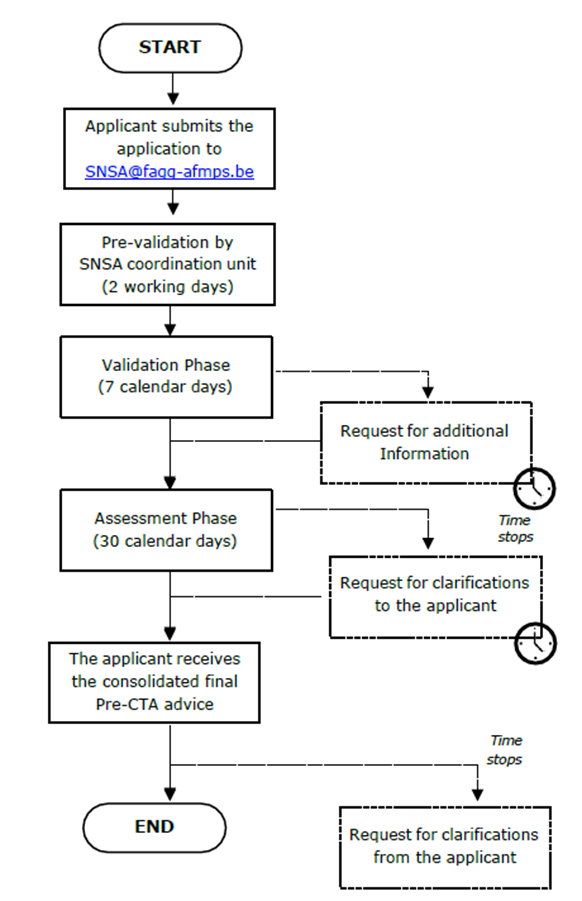
The developer should send the request for SNSA to the central contact point called “coordinating unit” (PEI/AEMPS/FAGG-AFMPS) with a draft list of questions raised using the SNSA application form and giving a brief outline of the scope of the SNSA request.
The list of question allows the designated regulators to start designating the appropriate experts for the request.
The brief outline of the scope of the request includes, for instance, drug product characteristics, manufacturing process for e.g. ATMP, background of the intended (pre) clinical development project, etc.)
Formal start of the procedure
The developer should send a cover letter, a detailed list of questions & clear position statement of the applicant for each question, the Briefing Document containing all detailed scientific and regulatory information regarding the dossier, and any additional supportive documentation.
The procedure starts when the briefing documents are sent.
Until day 20 minor updates can be made to the package if they have no impact on the choice of experts made.
Validation of the briefing document
There is a possibility of request of other information, clarification, or resubmission.
The procedure will formally start with the validation of the briefing document.
After validation, the SNSA meeting will take place at the latest in the following 55 days.
The SNSA meeting
It will be held virtually in most cases.
The lead agency will moderate the meeting.
No new questions or data can be added during the SNSA procedure or the meeting.
After the meeting
The leading authority will ask the Applicant to draft the meeting minutes of the formal SNSA meeting using the provided template.
The minutes should be sent to the lead and participating NCAs within 10 days after the meeting.
The consolidated advice will be sent to the applicant by day 90 of the procedure.
Within four weeks, the applicants should give feedback to the SNSA pilot to improve the questionnaire and the procedure.
Steps and timeline of the interaction, from EMA’s website
4. EMA-FDA Parallel Scientific Advice to interact with both the EMA in the European Union and the FDA in the United States of America
The EMA-FDA Parallel Scientific Advice aims to obtain feedback from both the EMA and the FDA on scientific issues and regulatory requirements.
It should be asked during the development phase of a medicine when both the European Union market and the United States market are targeted by the medicine’s developer for a future marketing authorisation and a potential commercialisation.
Who can request an EMA-FDA Parallel Scientific Advice?
The procedure is generally requested by a medicine developer on a voluntary basis. But the EMA and FDA may also initiate the EMA-FDA Parallel Scientific Advice process in full cooperation with the medicine developer in exceptional circumstances.
The medicine developer can be:
the “sponsor” of an Investigational New Drug Application in the United States,
the “applicant” that submits a New Drug Application or Biologics License Application in the United States,
or
a potential marketing authorisation applicant under the marketing authorisation process in the European Union.
Steps towards the Procedure
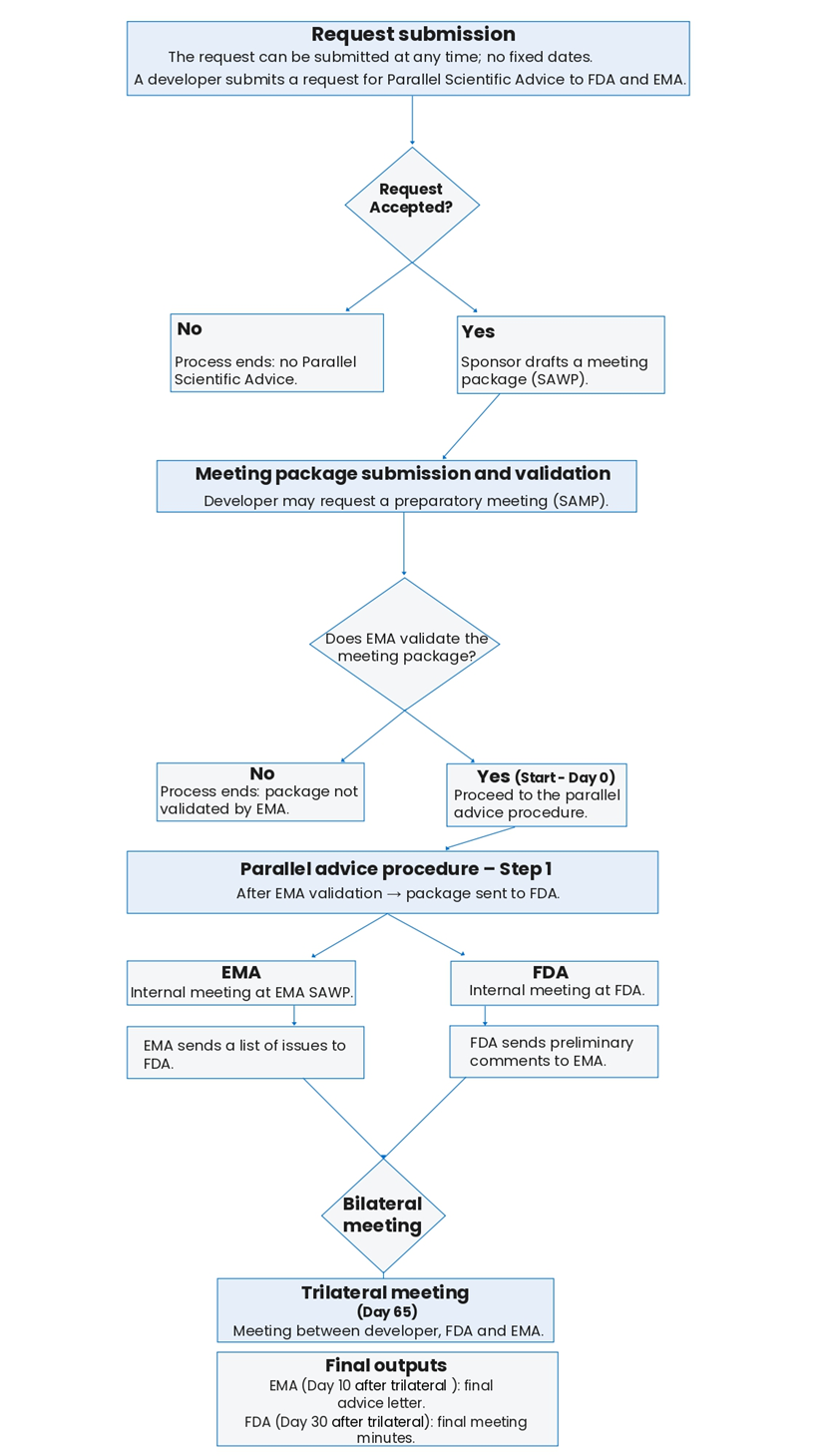
A developer can submit a request for a Parallel Scientific Advice to the FDA and EMA at any time, there is no fixed dates to request a parallel Scientific Advice.
A developer submits a request for a parallel scientific advice to the Food and Drug Administration (FDA) and to the European Medicines Agency (EMA).
Following this request, the agencies may accept or decline the request for Parallel scientific advice.
In the case of a negative answer from the agencies, the developer will not benefit from parallel scientific advice and the process ends there.
If the agencies accept the request, the developer will have to draft a meeting package accordingly to the SAWP procedures.
Meeting package submission and validation phase
During this stage of the procedure, it is possible for the developer to ask for a preparatory meeting according to the SAWP procedures.
Steps of the EMA-FDA Parallel Scientific Advice procedure initiated in the European Union
The procedure begins after EMA’s validation of the meeting package. It will last between 75 and 95 days. But, if EMA does not validate the meeting package, the procedure ends.
After EMA’s approval, the meeting package is sent to the FDA. An internal meeting is held at the FDA and another is held at the EMA SAWP.
The FDA sends preliminary comments on the meeting package to EMA, and EMA sends a list of issues to the FDA.
A bilateral meeting is held between the two agencies.
Then, a trilateral meeting between the developer, the FDA and EMA is organised during which the meeting package and the diverse issues related are raised and discussed.
Ten days after the trilateral meeting, the EMA adopts the final advice letter.
Thirty days after the trilateral meeting, the FDA adopts the final meeting minutes.
Please find below a flowchart on the EMA-FDA Parallel Scientific Advice procedure built from the EMA-FDA Parallel Scientific Advice Timetable available here.
Figure: Flowchart on the EMA-FDA Parallel Scientific Advice procedure
For more information on the timeline of the procedure, please see the document provided by EMA.
Joint Scientific Consultations (JSCs) and Parallel HTACG/EMA Joint Scientific Consultations
Applicable Articles related to HTACG joint scientific consultations and parallel HTACG/EMA joint scientific consultations : Articles 16 to 21 of Regulation (EU) 2021/2282 of the European Parliament and of the Council of 15 December 2021 on health technology assessment and amending Directive 2011/24/EU (HTA Regulation).
5. Joint Scientific Consultations (JSCs) to interact with all HTA bodies from the European Union via the HTA Coordination Group
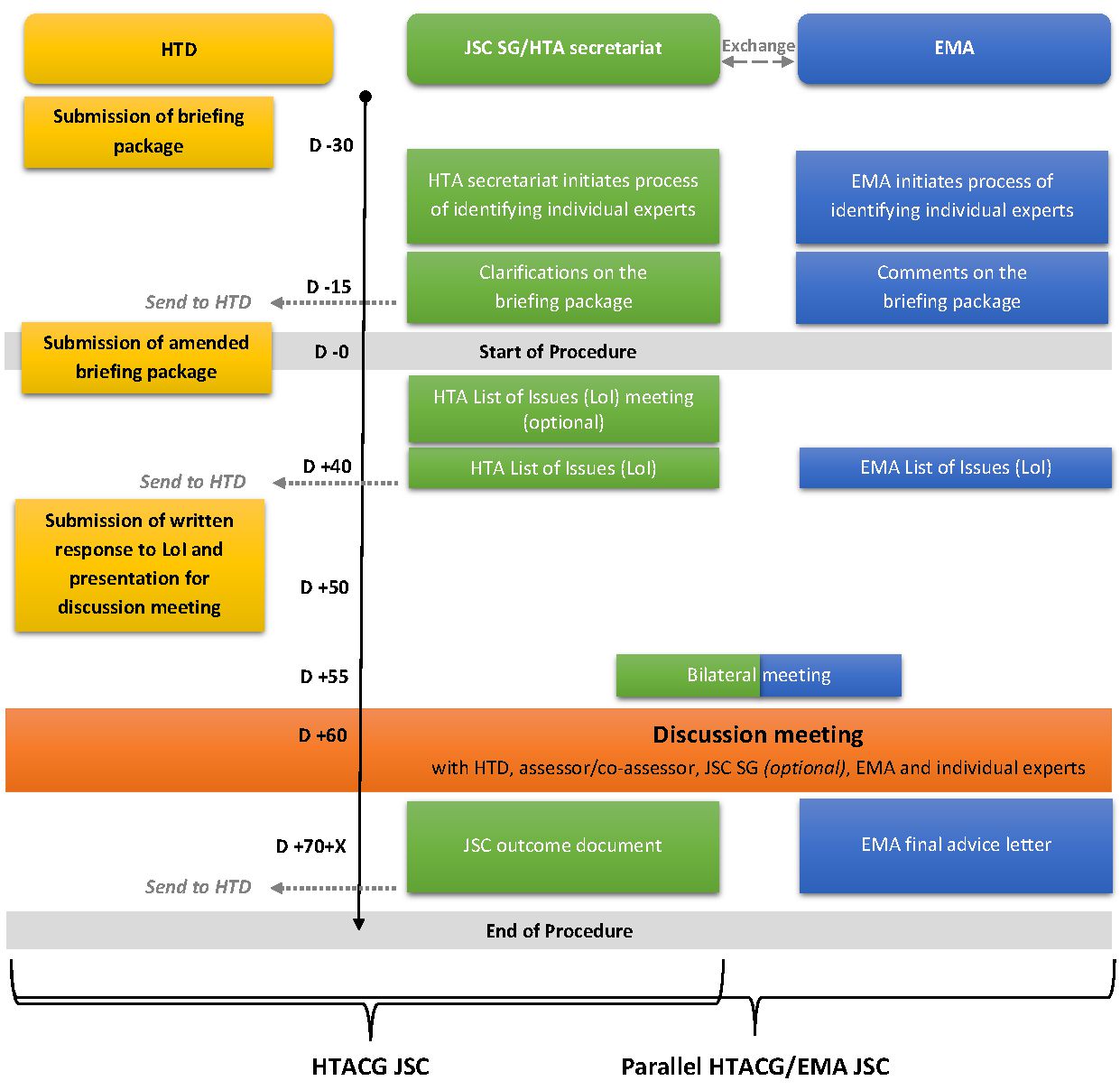
JSCs are coordinated by the Health Technology Assessment Coordination Group (HTACG) dedicated subgroup. The subgroup appoints assessors and co-assessors from different countries to monitor the process. The request can be for a new medicinal product or for an existing one but on a new indication.
Number of meetings per year
The Health Technology Assessment Coordination Group (HTACG) holds two quarterly meetings in the first half of the year and may increase the number to monthly meetings in the second half of the year (except in August). This possible increase depends on the number of JSCs initiated.
Request period for JSC
The request period and instructions on how to submit request is published on the European Commission website here and on the HTA IT platform.
In 2026, the periods are the following:
07 January to 04 February 2026
01 to 29 April 2026
03 June to 01 July 2026
23 September to 21 October 2026.
Aim of Joint Scientific Consultations (JSCs)
For Health Technology developers this procedure should be used to discuss the development plan of a health technology’s (here an ATMP) clinical development at an early stage. It will help developers to generate robust evidence.
“The Coordination Group shall carry out joint scientific consultations in order to exchange information with health technology developers on their development plans for a given health technology. Those consultations shall facilitate the generation of evidence that meets the likely evidence requirements of a subsequent joint clinical assessment on that health technology. […] Joint scientific consultations shall in particular concern all relevant clinical study design aspects, or clinical investigation design aspects, including comparators, interventions, health outcomes and patient populations” (Article 16(1) HTA Regulation).
Eligibility criteria and the selection criteria for JSCs for medicinal products
The eligibility of a product relies on two cumulative conditions:
The procedure will take approximately 4.5 months from the reception of the briefing document with annexes and references (briefing package) by the HTA secretariat via the HTA IT platform. At day 60 adiscussion meeting is held between the HTACG and the Health Technology developer.
6. Parallel Joint Scientific Consultations to interact with the HTA Coordination group and with the EMA (Parallel HTACG/EMA Joint Scientific Consultations)
Developers may request to enter into a Parallel HTACG/EMA Joint Scientific Consultations while in the process of asking for a Joint Scientific Consultation. This mechanism combines a Joint Scientific consultation from the HTA Coordination group with a Scientific Advice from the European Medicines Agency (EMA). This procedure has been established by article 17 of the HTA Regulation.
For this procedure the HTACG and the EMA processes run in parallel. Even though the two work separately they coordinate key steps of the procedure and share the necessary information to allow the scheme to work seamlessly. The timetable for JSCs will be synchronised with the timing of the process for Scientific Advice (SA). The questions asked by the developer in this procedure should be different from recent scientific advice already addressed by the EMA concerning a similar product.
Outcome of the scheme
The outcome will be a Scientific advice letter from EMA and a final JSC outcome document from the HTACG.
Joint scientific consultations at the HTACG and Parallel HTACG/EMA Joint Scientific Consultations
Regulation (EU) 2021/2282 of the European Parliament and of the Council of 15 December 2021 on health technology assessment and amending Directive 2011/24/EU (Text with EEA relevance)PE/80/2021/INIT CELEX number: 32021R2282
Commission Implementing Regulation (EU) 2024/3169 of 18 December 2024 laying down rules for the application of Regulation (EU) 2021/2282 of the European Parliament and of the Council with regard to the procedures for joint scientific consultations on medicinal products for human use at Union level, C/2024/8850 CELEX number: 32024R3169
Commission Implementing Regulation (EU) 2024/2699 of 18 October2024 laying down, pursuant to Regulation (EU) 2021/2282 of the European Parliament and of the Council, detailed procedural rules for the cooperation of the Member State Coordination Group on Health Technology Assessment and the Commission with the European Medicines Agency in the form of exchange of information as regards the joint clinical assessment of medicinal products and medical devices and in vitro diagnostic medical devices and as regards the joint scientific consultation on medicinal products and medical devices, C/2024/7201, Document 32024R2699, CELEX number: 32024R2699
Multistakeholder Advice at the European Medicines Agency: Is It Still Needed? - Vamvakas, Spiros, Jane Moseley, and Thorsten Vetter. Clinical Pharmacology & Therapeutics 105, no. 4 (2019): 819–821.
The New Regulation of Health Technology Assessments Reports - Wested, Jakob. European Health & Pharmaceutical Law Review (EHPL) 6, no. 3 (2022): 122–126.
International Harmonization for Cell and Gene Therapy Products - Arcidiacono, Judith. In Regulatory Aspects of Gene Therapy and Cell Therapy Products, 235–240. Springer, Cham, 2023.
Incorporating the Patient Experience into Regulatory Decision Making in the USA, Europe, and Canada - Kluetz, Paul G, Daniel J O’Connor, and Katherine Soltys. The Lancet Oncology 19, no. 5 (1 May 2018): e267–274.
Multistakeholder Advice at the European Medicines Agency: Is It Still Needed? - Vamvakas, Spiros, Jane Moseley, and Thorsten Vetter. Clinical Pharmacology & Therapeutics 105, no. 4 (2019): 819–821.
Annual Work Programme 2025, Adopted on 28 November 2024, by the HTA CG pursuant to Article 6 of Regulation (EU) 2021/2282 on Health Technology Assessment, V1.0, 28 November 2024, HTACG