Any company wishing to market Advanced Therapy Medicinal Products (ATMPs) within the European Union must hold a marketing authorisation issued by the European Commission.
Different options are offered by the European Union for being granted a marketing authorisation through specific regulatory mechanisms to facilitate early patient access to medicinal products, including ATMPs, while maintaining a high assessment of the quality of these medicines, and of their safety and efficacy regarding the benefit-risk balance.
The accelerated assessment is a procedural tool to reduce the centralised procedure review period of a marketing authorisation applicant from 210 to a maximum of 150 days, excluding clock stops. An applicant may request an accelerated assessment for medicinal products if they are of major public health interest, in particular from the viewpoint of therapeutic innovation. The request shall be duly substantiated and its acceptance will be decided by the Committee for Medicinal Products for Human Use (CHMP). (Article 14 § 9 of Regulation (EC) No 726/2004). Nevertheless, the decision on accelerated assessment has no impact on the future CHMP opinion on whether a marketing authorisation should be granted.
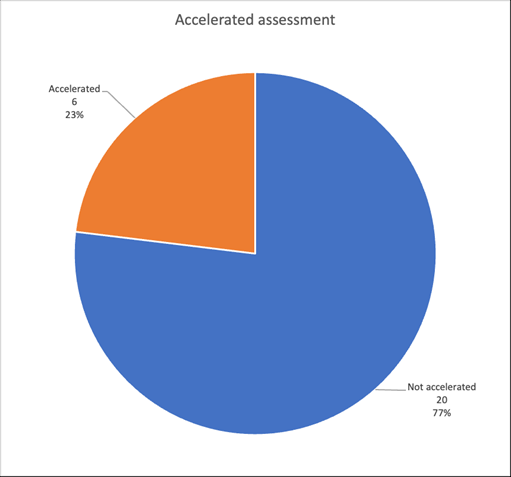
The accelerated assessment is particularly adapted to ATMPs, one of whose characteristics is to be innovative (Fig. 1).